Сайты связывания транскрипционного фактора в участке хромосомы человека
Проверка качества ридов
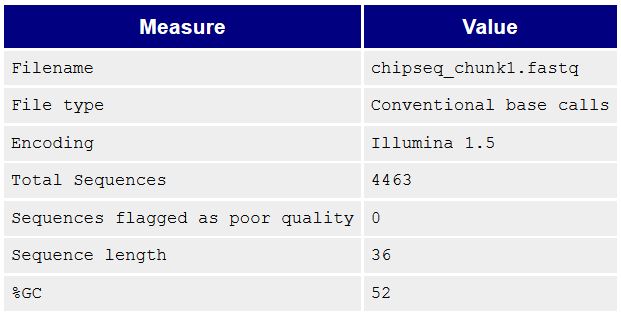
Был дан файл chipseq_chunk1.fastq.Сперва нужно было проверить, необходима ли очистка файла. Для этого была использована программа FastQC.
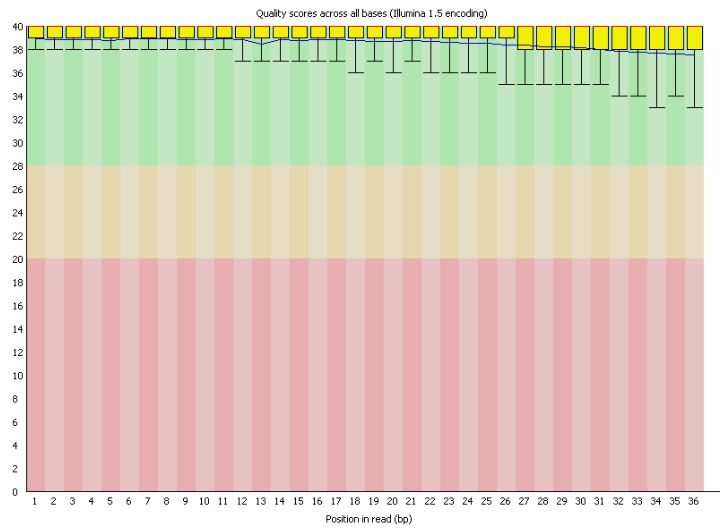
В реультате на выходе был получен html-файл. Видно, что риды очень высокого качества, поэтому никакой чистки не нужно.
{kind=link}
*Ссылка на изображение с общей статистикой по ридам в fastq-файле.
{kind=link}
Картирование ридов на геном человека
Картирование ридов было выполнено при помощи BWA, при этом референс индексированный:
bwa mem /srv/databases/ngs/hg19/GRCh37.p13.genome.fa chipseq_chunk1.fastq > chipseq_chunk1.sam
Для анализа картирования предложено было использовать следующую последовательность команд. Использую пакет samtools, sam-файл был переведен в файл бинарного формата, bam-файл:
samtools view chipseq_chunk1.sam –bSo chipseq_chunk1.bam
Далее для сортировки выравнивания ридов с референсом по координате в референсе была использована следующая команда:
samtools sort chipseq_chunk1.bam -T chip_temp -o chipseq_chunk1_sort.bam
И после проиндексирована:
samtools index chipseq_chunk1_sort.bam
Наконец для получения статистики по картированию:
samtools idxstats chipseq_chunk1_sort.bam > chip_out.txt
Видно (chip_out.txt файл), что все 4463 рида были картированы на геном, причем более 90% (90.57%) картированы на 12 хромосому. Поэтому, судя по выдаче, риды именно с 12 хромосомы предложены для анализа.
Поиск пиков
Для поиска пиков была использована программа MACS. Так как пиков было слишком мало, пришлось использовать следующую команду (присутствует флажок --nomodel):
macs2 callpeak -n chipseq_chunk1 -t chipseq_chunk1_sort.bam --nomodel
В результате были получены три файла (narrowPeak, xls, bed), в каждом из которых содержится информация о пиках.
Всего было найдено 5 пиков на 12 хромосоме (Таблица 1). Видно, что чем больше -log10(p-value), тем меньше p-value, а значит достоверность находки выше. У абсолютно всех находок p-value достаточно низкий.
| Таблица 1. Характеристика пиков | ||||||
|---|---|---|---|---|---|---|
| Номер пика | Начальная позиция в геноме, пн | Конечная позиция в геноме, пн | -log10(p-value) | Пиковая позиция, пн | Расстояние от начальной позиции до пиковой, пн | Расстояние от пиковой позиции до конечной, пн |
| 1 | 132061389 | 132061963 | 33.741 | 132061552 | 163 | 411 |
| 2 | 132354625 | 132354910 | 22.122 | 132354760 | 135 | 150 |
| 3 | 132628815 | 132629113 | 33.726 | 132628971 | 156 | 142 |
| 4 | 132970034 | 132970233 | 15.474 | 132970148 | 114 | 85 |
| 5 | 132970737 | 132970944 | 13.145 | 132970830 | 93 | 114 |
Для визуализации пиков использовалась программа UCSC Genome Browser. На вход подавался этот файл, который отличается от обычного наличием двух строчек в начале:
track type=narrowPeak visibility=3 db=hg19 name="my_peaks" description="Peaks from chunk 1"
browser position chr12:132061380-132970950
Результат виден на картинке ниже.
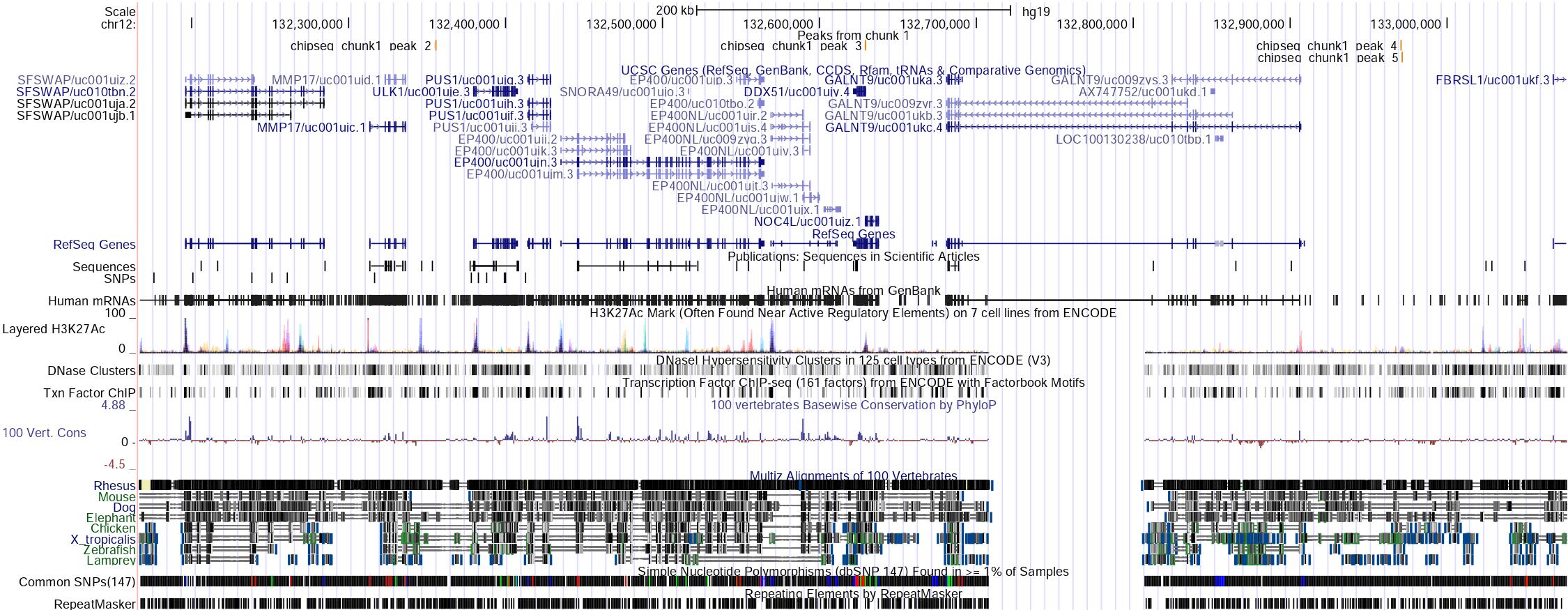
Интересно, что все пики не соответствуют кодирующей части гена. Однако 3 пик пересекается с функциональным элементом генома. Именно поэтому решено было посмотреть на него вблизи. Изображение представлено внизу.

Пик 3 располагается между двумя экзонами генов (uc001ujy.4 и uc001ujz.1), причем пиковая часть не пересекается с кодирующими частями генов. Расстояние от пиковой позиции до промотора составляет примерно 10-15 пн. Скорее всего этот ТФ в этом месте оказывает влияние на предпромоторную часть гена uc001ujz.1.
Что касается остальных пиков, то тут наблюдается другая картина. Расстояние от пикового значения до ближайших кодирующих частей генов гораздо больше - сотни, тысячи пн. Возможно ТФ таким образом в этих сайтах регулирует работу энхансеров (как пример).
Далее была использована команда для получения fasta файла:
/srv/databases/ngs/tools/seqtk/seqtk subseq /srv/databases/ngs/hg19/GRCh37.p13.genome.fa
chipseq_chunk1_peaks.narrowPeak > my_peaks.fa
Последовательности из fasta файла были выровнены в Jalview. Результат представлен ниже (окарска по степени консервативности):

Видно, что в выравнивании большое количество гэпов, при этом количество консервативных позиций невелико. Поэтому определить консенсусную последовательность длиной 4-8 пн невозможно.
⌘
© Emir Radkevich, 2016