Сборка генома de novo
Задание 1
Мне был выдан SRR4240359 — определённый код доступа проекта по секвенированию генома Buchnera aphidicola.
Это вид гамма-протеобактерий, являющихся первичными эндосимбионтами гороховых тлей Acyrthosiphon pisum, основным источником пищи которым служат соки растений. При этом у тлей возникает проблема сбалансированности рациона: углеводов они получают предостаточно, в то время как аминокислот нехватает. Поэтому у тлей примерно 200 миллионов лет назад возник симбиоз c Buchnera. Эти бактерии в результате таких отношений потеряли большую часть своего генома, в частности, у них теперь отсутствуют многие ферменты, которые есть у свободно живущих бактерий. Живут они в определённых клетках тли — бактериоцитах — и синтезируют незаменимые аминокислоты, в которых тли как раз и нуждаются. В ответ же сами бактерии получают от хозяина множество питательных веществ, а также среду, богатую азотом, которая необходима для самого важного — синтеза аминокислот. Без хозяина бактерии не могли бы этим заниматься, что связано со значительным уменьшением генома. Передаются симбионты вертикально: через яйцеклетки матери к потомкам, что обеспечивает непрерывное "заражение". В одной особи тли может существовать одновременно до 5.6 × 106 бактерий Buchnera[1][2][3].
Работа с данной бактерией может дать возможность узнать больше о молекулярных основах внутриклеточного эндосимбиоза и о паразитическом поведении симбионтов.
Предлагаю посмотреть интересное видео от людей, изучающих эти симбиотические отношения.
Итак, я обработала файл [fastq] c чтениями (тип секвенирования: Illumina) программой Trimmomatic. Были удалены адаптеры: в файле adapters.fasta я собрала все известные адаптеры, и передала его в параметре обработки ILLUMINACLIP. Также были обрезаны концы ридов с неудовлетворительным качеством: TRAILING:20, и отсечены чтения малой (< 30) длины: MINLEN:30. Полная команда:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 SRR4240359.fastq SRR4240359_trimmed.fastq TRAILING:20 MINLEN:30 ILLUMINACLIP:adapters.fasta:2:7:7
Выдача программы на stdout следующая:
Input Reads: 13557938 Surviving: 12562254 (92,66%) Dropped: 995684 (7,34%)
Таким образом, обработка привела к потере почти миллиона ридов, что составило ~7% от изначального их количества. При этом размер файла сократился с 1375Mb (1.34Gb) до 1257Mb (1.23Gb). Таким образом, размер сократился на 8.2% процента. Держа в памяти, что речь идет о гигабайтах информации, понятно, что это неплохой результат чистки. Но и сработала она, надо сказать, не мгновенно.
При нашей длине чтения максимальным значением для k-меров будет k=29. Тип чтений в нашем [fastq]-файле: короткие непарные: short.
Для подготовки k-меров использовалась программа velveth[4]. Она принимает на вход последовательности и создаёт хэш-таблицу, в результате выдаёт два файла (Sequences и Roadmaps), необходимые для работы с программой velvetg. Длина k-мера, или длина хэша, соответствует длине хэшируемого слова в парах оснований. В задании она была указана. Но вообще выбор этого параметра это отдельная задача, которая требует учёта следующих технических ограничений:
- во избежание палиндромов
kдолжно быть нечётным; k ≤ 31, где 31 — дефолтная максимальная хэш-длина (этот параметр, впрочем, может быть сменен вручную);kстрого меньше длины ридов, нарушение чего очевидно приводит к последующему отсутствию перекрываний.
Выбирая длину k-мера, нужно балансировать между большими и маленькими значениями. Большие значения, несомненно, избавят от возникающих ложных перекрываний ридов, но уменьшится общее покрытие. С малыми значениями — наоборот.
Итоговая команда запуска velveth:
velveth pr15 29 -short -fastq SRR4240359_trimmed.fastq
Примечание: pr15 — директория, в которую записывались выходные файлы.
Сборка на основе k-меров производилась при помощи программы velvetg, которая строит и анализирует граф де-Брёйна. В нём k-меры являются вершинами, а ориентированные рёбра соединяют так называемые префикс- и суффикс-последовательности.
При первом запуске авторы рекомендуют указывать параметр порогового покрытия так: -cov_cutoff auto. Значение auto рассчитывается как половина от длины взвешенной медианы по глубине покрытия контига. Итоговая команда:
velvetg pr15 -cov_cutoff auto
Результаты работы velvetg
- изначально 1 850 763 вершин, в финальной версии графа — 87;
N50 =120256;- файлы с графами:
PreGraph, LastGraph, Graph; stats.txt— информация о вершинах графа:- длина вершины в k-мерах (для расчёта длины в нуклеотидах нужно просто добавить
k–1); in— число входящих рёбер с 5'-конца;out— число выходящих рёбер с 3'-конца;- колонки
*_covи*_Ocovнесут информацию о покрытии, различаются между собой методом вычислений (*_Ocov— более строгие расчёты).
- длина вершины в k-мерах (для расчёта длины в нуклеотидах нужно просто добавить
contigs.fa— [fasta]-файл с контигами — последовательностями длины не менее2k(мы не устанавливали специальное ограничение на минимально возможную длину контига, но такая возможность, вообще говоря, существует).- анализ контигов удобнее проводить по
stats.txt: конкретные нуклеотидные последовательности нас пока не особо интересуют. При этом надо учитывать, что не все вершины стали контигами (всего их 48 штук, а вершин, как было сказано ранее, 87). Нужно сперва отсечь все те, длина которых меньше 29, ибо они не прописываются вcontigs.faи являются "неполноценными контигами". - данные по контигам:
- среднее значение длины контигов (в k-мерах): 13702.88;
- медианное значение длины: 112.5;
-
самые длинные контиги:
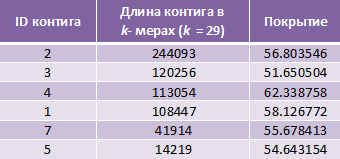
- среднее значение покрытий: 56.72;
- медианное значение покрытий: 55.07;
-
контиги с наибольшим и наименьшим покрытием (аномальными по сравнению с медианным значением их всё же сложно признать):
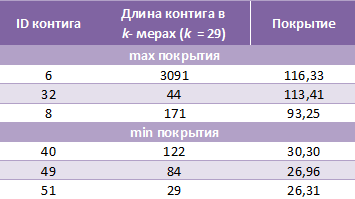
Анализ
Для анализа были взяты три самых длинных контига (ID2, ID3, ID4). Они были по отдельности переданы на обработку алгоритмом megablast в паре с хромосомой B. aphidicola (GenBank AC: CP009253). Результаты всех трёх запусков приведены ниже.
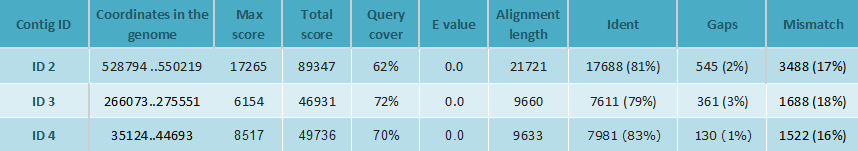
Аналогичный анализ для контигов с аномальными покрытиями проведён не был, поскольку, как было сказано, таких у меня просто не получилось.
Задание 2*
Проблема выбора длины хэшируемого слова описана выше. В этом задании предлагалось повторить работу при k=25. В таблице ниже указаны основные параметры, по которым можно сравнить полученные контиги.
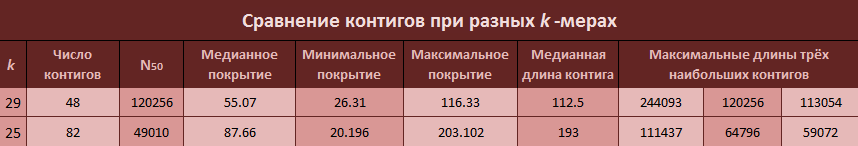
Результаты вполне ожидаемые. При уменьшении хэш-длины соответственно уменьшаются и контиги, а их покрытие — растёт. Единственным расхождением с ожиданием стала медианнаядлина контига. Тут она почему-то получилась выше. Это может быть связано со значительным увеличением числа контигов.
Для трёх самых длинных контигов, как и в предыдущем случае, был запущен megablast с последовательностью хромосомы изучаемой бактерии. Соответствующие последовательности лежат в директории /nfs/srv/databases/ngs/emkeller/pr15/25/. Результаты приведены ниже.
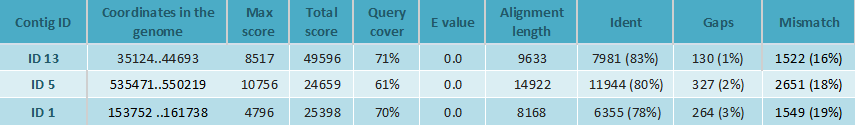
Стоит отметить, что участок хромосомы в выравнивании с ID13 соответствует выравниванию в предыдущем случае с ID4, а выравнивание с ID5 соответствует выравниванию c прошлым ID2. Третье же перекрывание, с ID1, не перекрывается ни с каким из ранее полученных.
Задание 3*
Программа SPAdes[5] изначально задумывалась как сборщик небольших геномов и тестировалась на заданиях по сборке геномов бактерий и грибов. Перед сборкой рекомендовано проводить исправление ошибок в ридах при помощи BayesHammer[6], алогритма, который по умолчанию встроен в пайплайн SPAdes. Он основывается на сочетании алогритма Байесовской кластеризации и построении графов Хэмминга. Вершинами в таких графах являются k-меры, а каждое ребро обладает весом — числом, равным так называемому расстоянию Хэмминга. Оно определяется как количество позиций, в которых риды различны между собой. Ребро проводится, если его вес не превышает заданный порог (т.е. последовательности "несильно" отличаются друг от друга).
При построении графа алгоритм HAMMING выделяет определенные кластеры связности. Но такая кластеризация сильно зависит от равномерности покрытия. Чаще всего покрытие неравномерное, и из-за этого могут возникать дополнительные ошибки. Для того чтобы этого избежать и применяется субкластеризация по Байесу: она улучшает построенные на предыдущем шаге кластеры, учитывая качество прочтения в ридах. Затем в каждом отдельном кластере выбирается "центральная" вершина, которая считается в дальнейшем эталоном — программа определяет её как реально пристуствующую в геноме, а все связанные с ней объявляются "ошибочными". Исправление происходит сперва на уровне k-меров, затем и в самом риде, что повышает точность последующей сборки.
К сожалению, ограничение в памяти не позволило мне запустить SPAdes в режиме с коррекцией. Более того, программа по умолчанию выполняет сборки при различных значениях k. Но нужно понимать, что это очень времязатратный процесс. Поэтому, хоть это и нарушает исходно задуманный ход работы программы, мне пришлось ограничиться одним значением и установить его вручную. Я выбрала k=29 для наглядности в сравнении с работой velvet.
Итоговая команда запуска:
-m 500 -s SRR4240359_trimmed.fastq --only-assembler -k 29 -o spades
Таким образом, сборка проводилась по улучшенным при помощи Trimmomatic ридам (-s задаёт формат ридов во входном файле, в данном случае — непарные). Выходные файлы складывались в директорию spades.
Параметр -m 500 устанавливает предел использования памяти в гигабайтах.
По умолчанию это значение равняется 250Gb, но при таких, казалось бы, щедрых условиях, на определенном этапе программа, не окончив работу, выводила сообщение об ошибке и нехватке памяти. При этом не спасало и
отключение модуля по исправлению ошибок. В итоге всё заработало только при указаном лимите в 500Gb и выполнении исключительно сборки (параметр --only-assembler).
Ниже приведена выдержка из log-файла работы SPAdes.
Output dir: /nfs/srv/databases/ngs/emkeller/spades
Mode: ONLY assembling (without read error correction)
Debug mode is turned OFF
Dataset parameters:
Multi-cell mode (you should set '--sc' flag if input data was obtained with MDA (single-cell) technology
Reads:
Library number: 1, library type: single
left reads: not specified
right reads: not specified
interlaced reads: not specified
single reads: ['/nfs/srv/databases/ngs/emkeller/SRR4240359_trimmed.fastq']
Assembly parameters:
k: [29]
Mismatch careful mode is turned OFF
Repeat resolution is enabled
MismatchCorrector will be SKIPPED
Other parameters:
Dir for temp files: /nfs/srv/databases/ngs/emkeller/spades/tmp
Threads: 16
Memory limit (in Gb): 500
Удивительно, но даже при таких мягких параметрах программа работала 2 часа 50 минут. На выходе были получены среди прочих следующие файлы: contigs.fasta, contigs.fastg
(файлы с последовательностями контигов и информацией о них); scaffolds.fasta, scaffolds.fastg (аналогично для скэффолдов).
Из файла contigs.fasta была извлечена информация о длине и покрытиях отдельных контигов.
Соответствующий скрипт доступен для скачивания. Выходной файл после работы скрипта: output.txt был обработан средствами MS Excel. Получившуюся таблицу со сравнением основных параметров сборки программами velvet и SPAdes можно видеть ниже.
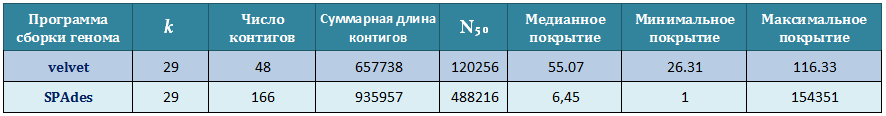
Стоит отметить, что основной параметр, по которому мы оцениваем качество сборки — N50 — повышается, а также суммарная длина контигов возрастает, что говорит о повышении качества сборки.
В этом случае у нас появились аномальные значения покрытий. Я взяла контиг с максимальным покрытием, указанным в таблице, но даже не стала пробовать запускать megablast. Достаточно посмотреть на последовательность этого "аномального" контига, чтобы понять, что в этом нет смысла.
>NODE_160_length_32_cov_154351_ID_319 ACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
Контиг с наименьшим покрытием величиной 1.00 тоже можно счесть аномальным. Правда, как мы видим, он состоит из повторов и относится к так называемым low complexity regions. И даже при отключении фильтрации таких участков, которая по умолчанию происходит при запуске megablast, результаты не были найдены.
>NODE_146_length_34_cov_1_ID_291 AATGAAATGAAATGAAATGAAATGAAATGAAATG
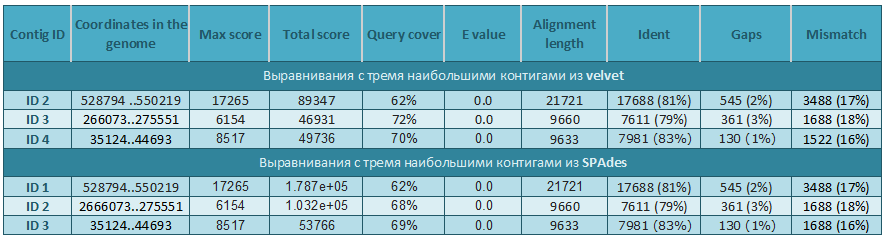
Выравнивания трёх самых длинных контигов получились аналогичны тем, что получались с контигами из velvet.
[1] — Microbe Wiki: B.aphidicola
[2] — Microbe Wiki: Aphids and Buchnera
[3] — Wiki: B. aphidicola
[4] — Manual: velveth
[5] — Manual: SPAdes
[6] — BayesHammer