Выравнивание геномов и EMBOSS
Задание 1. Упражнения по EMBOSS
Упр. 1
Задание: Транслировать данную нуклеотидную последовательность в шести рамках.
Входные данные: my.fasta — нуклеотидная последовательность.
Команда:
transeq my.fasta -frame 6 -out mytransl.fasta
Результат: mytransl.fasta — файл с шестью последовательностями аминокислот, получающихся в ходе формальной трансляции с использованием шести возможных рамок считывания.
Упр. 2
Задание: Перевести аннотации особенностей в записи формата [gb] в табличный формат [gff].
Входные данные: chr.gb — genbank-последовательность хромосомы
Команда:
featcopy chr.gb -out chr.gff
Результат: chr.gff — файл с особенностями в табличном [gff]-формате.
Упр. 3
Задание: Из данного файла с хромосомой в формате [gb] получить [fasta]-файл с кодирующими последовательностями и добавить в описание каждой последовательности функцию белка.
Входные данные: chr.gb — genbank-последовательность хромосомы.
Команда:
extractfeat chr.gb -out cdsinchr.fasta -type CDS -describe product
Результат: cdsinchr.fasta — файл с кодирующими последовательностями. В описании последовательностей указан продукт в формате (product="...").
Упр. 4
Задание: Найти частоты динуклеотидов в данной нуклеотидной последовательности и сравнить их с ожидаемыми.
Входные данные: my.fasta — нуклеотидная последовательность.
Команда:
compseq my.fasta -out my_dinucl.txt -word 2
Результат: my_dinucl.txt — файл с наблюдаемыми и ожидаемыми частотами появления нуклеотидов.
Упр. 5
Задание: Выровнять кодирующие последовательности соответственно выравниванию белков - их продуктов.
Входные данные: nuclseqs.fasta — файл с кодирующими последовательностями, protalign.fasta — файл с выравниванием белков-продуктов.
Команда:
tranalign nuclseqs.fasta protalign.fasta -out nuclalign.fasta -table 4
Параметр -table был указан для корректной трансляции генетического кода, которая имеет свои особенности в различных таксонах. Соответствующий параметр можно узнать из квалификатора '/transl_table'.
Результат: nuclalign.fasta — файл с выравниванием кодирующих (нуклеотидных) последовательностей.
Задание 2(б). Построение нуклеотидного пангенома при помощи NPG-explorer
Для построения нуклеотидного пангенома я выбрала три штамма Staphylococcus aureus (MRSA252, Mu50, JH1).
Рис. 1. Staphylococcus aureus
Staphilococcus aureus[1] — грамположительная шаровидная бактерия из типа Firmicutes, которая зачастую обнаруживается в дыхательных путях и на коже. Является факультативным аэробом и может существовать в условиях отсутствия кислорода. Несмотря на то, что S. aureus не всегда является патогеном, он всё же признан одной из самых распространенных причин абсцессов, респираторных инфекций (таких как, например, синусит) и отравления еды. Патогенные штаммы зачастую вызывают инфекции путём синтеза белков-токсинов и мембранных белков, которые связываются с антителами и инактивируют их. Существование устойчивых к антибиотикам штаммов S. aureus (например, MRSA — methicillin-resistant S. aureus) представляет собой серьёзную проблему, борьба с которой продолжается и по сей день.
Построение нуклеотидного пангенома
Для построения использовалась программа NPG explorer на kodomo.
Сперва я создала рабочую директорию, которая доступна по следующему адресу:
emkeller/term3/block2/npg
В ней создала файл genomes.tsv, который содержал в определенном формате ссылки на геномные последовательности трёх выбранных мной штаммов.
Затем выполнила следующие команды для подготовки к созданию пангенома и получения рекомендуемой оценки параметра MIN_IDENTITY (это значение минимального порога для доли консервативных позиций во всех блоках, кроме минорных m-blocks). После чего создала файл конфигурации npge.conf, в котором для этого параметра сменила значение с умолчательного 0.9 на то, что было несколько ниже рекомендованного (0.683) (0.883 из /examine/identity_recommended.txt).
npge Prepare npge Examine npge -g npge.conf
После чего было запущено создание пангенома, а выдача протокола выполнения перенаправлена в файл log:
npge MakePangenome > log
После того, как программа закончила свою работу, нужно было ввести ещё одну команду для создания файлов, содержащих информацию о построенном пангеноме:
npge PostProcessing
Описание синтеничных участков
В данном задании были проанализированы синтеничные участки, или так называемые глобальные g-блоки.
в папке /global-blocks лежат файлы которые были получены в результате пост-процессинга и содержат ценную информацию как о самих блоках (blocks.gbi), так и о их расположении на хромосомах отдельных штаммов и в итоговой пангеномной последовательности (blocks.blocks).
Вся извлечённая и отформатированная информация (в данном и последующих заданиях) содержится в [xlsx]-файле и доступна для скачивания.
g-блоки состоят из последовательно идущих во всех блоках s-блоков, между которыми разрешено наличие блоков других типов (r/h/u/m). Важно отметить то, что во всех файлах, связанных с глобальными блоками, появляются и проанализированы i-блоки. Этим как раз обозначаются блоки всех остальных (не s) типов. Для данного задания было решено удалить из выравнивания строки, не содержащие g-блоков.
Всего g-блоков 35, из которых 24 оказались консервативными и присутствовали во всех трех геномах на одной и той же позиции в выравнивании.
Из выравнивания (Рис. 2) хорошо видно, что последовательности g-блоков JH1 и Mu50 близки между собой и отличаются от MRSA252, ибо есть немало позиций, в которых у MRSA252 стоит прочерк (гэп), а у JH1 и Mu50 — какой-то g-блок. И, соответственно, поскольку блоки есть во всех трёх последовательностях, есть и обратные ситуации: у MRSA252 стоит какой-то блок, а у двух других последовательностей в этой позиции прочерки. Таким образом, речь идёт о транслокации некоторых блоков. Характер перемещения таков, что обычно сдвиг у MRSA252 происходит на две позиции, что может быть объяснено кольцевой формой хромосомы. На примере g3x108 можно заметить, что иногда в этих случаях у MRSA252 направление соответствующего блока противоположно направлению блоков в двух других геномах, т.е. произошла инверсия.
Рис. 2. Выравнивание g-блоков

Отметим, что есть и такие ситуации, в которых отличаются JH1 и Mu50. Так, блок g3x593 у JH1 встречается в одной позиции, в то время как у двух оставшихся он стоит в другой позиции. (Здесь тоже можно заметить смену направления блока.)
В числе прочих можно отметить такую особенность, как наличие вставки i-блока i2x2 у JH1 и Mu50 (у MRSA252 в этой позиции находится g-блок.
Описание ядра геномов
Ядро геномов составляют s-блоки. Это фрагменты, присутствующие во всех геномах. Информация о них взята в файле /pangenome/pangenome.info и теперь доступна по ссылке.
- Число s-блоков: 579
- Суммарная длина (процент от средней длины генома): 2 455 619 (82.38%)
- Сходство геномов (доля консервативных позиций в объединенном выравнивании s-блоков): 0.9762
Описание повторов
r-блоки — это блоки с повторами хотя бы в одном геноме. Общая информация:
- Число r-блоков: 460
- Суммарная длина (процент от средней длины генома): 132 203 (4.43%)
- Доля консервативных позиций в объединенном выравнивании r-блоков: 0.8246
Более подробная информация была извлечена из /pangenome/pangenome.bi и извлечена в ранее указанный [xlsx]-файл, после чего созданы листы с информацией по каждому типу блоков, в том числе и по r-блокам.
Также информацию по r-блокам можно просмотреть на Рис. 3.
Рис. 3. Информация по r-блокам
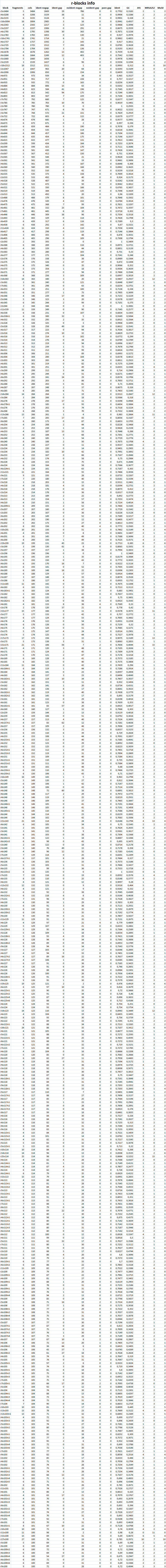
Рассмотрим блок r6x3208, второй по длине из имеющихся (3208 нуклеотидов), имеющий по 2 повтора в каждом геноме. Процент консервативности (ident) данного блока составляет 99%, что в совокупности со сказанным ранее позволяет предположить наличие в блоке кодирующего участка.
Действительно, qnpge сообщает о наличии 24 генов в этом блоке (сумма по геномам).
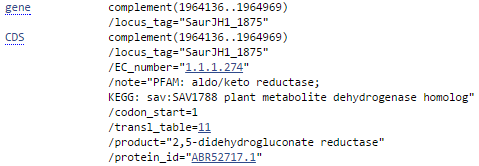
Ещё один интересный блок — это, например, r21x143. Он обладает сравнительно небольшой длиной, но значительным числом фрагментов (21), из которых 13 принадлежат MRSA252. Стоит отметить, что все гены за исключением одного аннотированы как транспозазы (иногда предполагаемые). Ген-исключение принадлежит JH1. Он аннотирован как 2,5-дидегидроглюконат редуктаза. Соответствующее подтверждение из reference sequence:
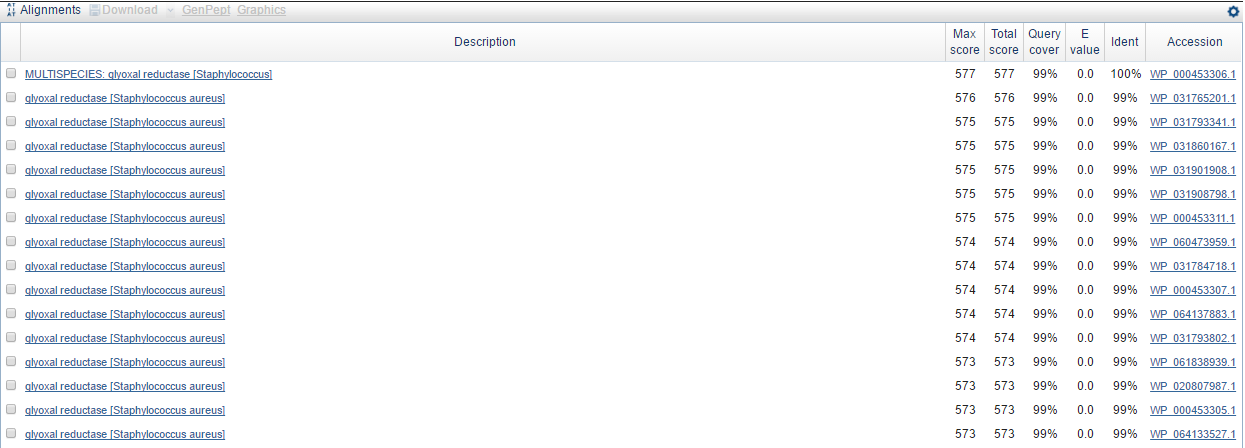
Можно окончательно убедиться в том, что, несмотря на обилие транспозаз-кодирующих генов в исследуемом блоке, выбранный ген действительно выполняет указанную в аннотации функцию. Для этого его последовательность была прогнана через blastx. Лучшие находки представлены на рисунке ниже. Поскольку практически все находки имели E-value 0.0, Query cover 99-100% и принадлежали глиоксаль-редуктазам S.aureus, то можно сделать вывод о том, что аннтоация была правильной.
Описание крупных делеций
h-блоки — это так называемые полустабильные блоки, состоят из фрагментов, содержащихся не во всех, а лишь в части геномов. Общая информация:
- Число h-блоков: 172
- Суммарная длина (процент от средней длины генома): 153 446 (5.14%)
- Доля консервативных позиций в объединенном выравнивании h-блоков: 0.9868
Подробную информацию по h-блокам можно просмотреть на Рис. 4.
Рис. 4. Информация по h-блокам
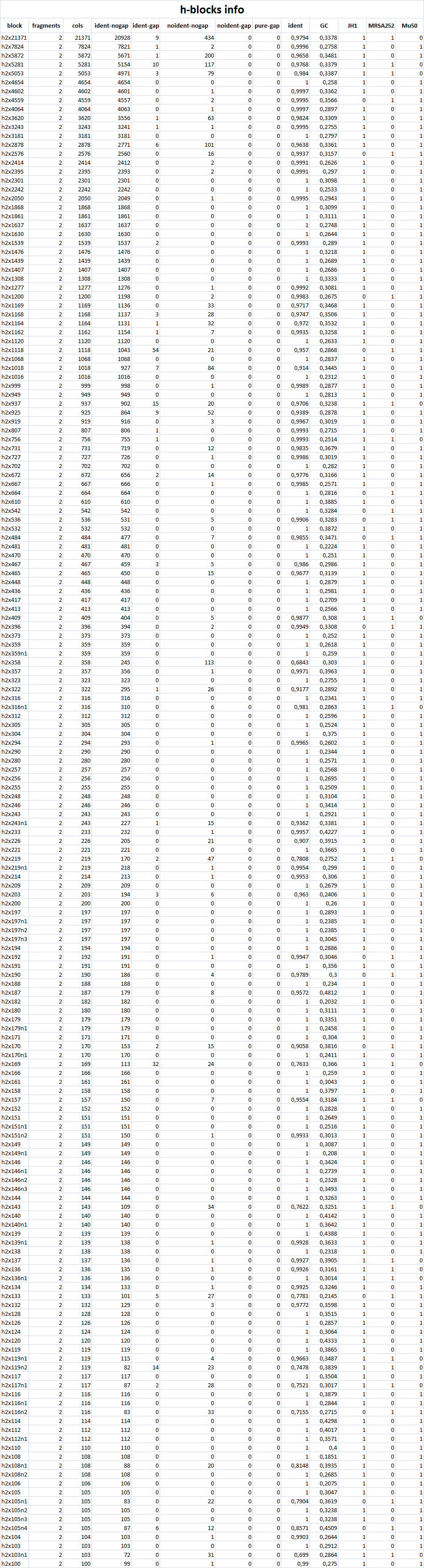
Сводка из qnpge для двух h-блоков:
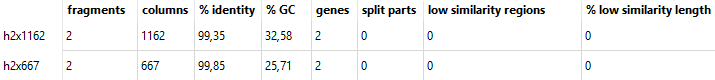
Оба блока, как следует из Таблицы 4, не встречаются у MRSA252, что говорит о достаточно крупных делециях. Интересно узнать, что кодировали гены, которые были в этих блоках:
- h2x1162: excisionase (JH1); integrase (Mu50)
- h2x667: type I restriction-modification system, M (JH1); probable specificity determinant HsdS (Mu50)
Но на самом деле, хоть речь и идет о делеции, бесследно белки не пропали. Для одного из них, белка субъединицы М системы рестрикции-модификации было проверено его наличие в геноме MRSA252. И он там есть, просто кодируется совершенно в другом участке генома.
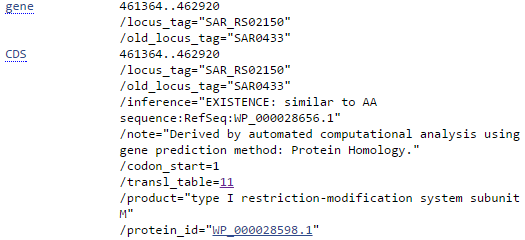
Напоследок замечу, что всё же смело говорить тут нельзя: обоснование для такой аннотации было, как указывает квалификатор '/note', получено в ходе автоматизированного предсказания генов. Возможно, не стоит этому слишком сильно доверять.
Примеры расхождений между аннотациями генов одного блока
Проблема 1. В блоке h2x7824 на одном из участков выравнивания у Mu50 не распознан ген, хотя последовательность в этой области абсолютно аналогична JH1.

Проблема 2. В блоке s3x950 с 354 позиции в каждом из трех геномов начинается участок гена. (Cм. рисунок ниже) Сходство последовательностей на этом участке практически 100%. Соответственно, ожидаем, что и белок, закодированный в этих генах, будет один и тот же. Но npge выдаёт, что у MRSA252 и Mu50 это субъединица А гиразы ДНК, а у JH1 — это карбогидрат киназа, YjeF-related protein.

При обращении к геному JH1 находим следующее:
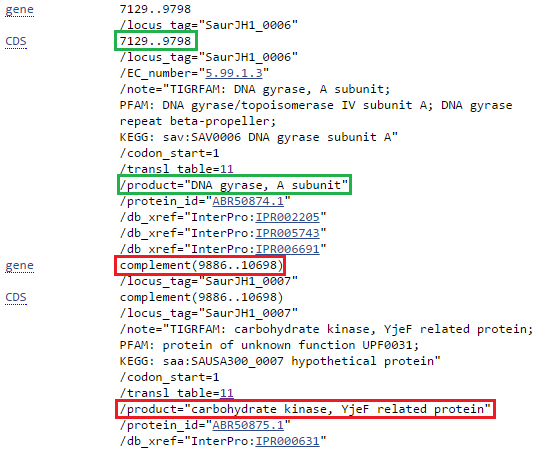
Красным я выделила то, что выдаёт npge. Но можно заметить, что координаты CDS совершенно не те, что на самом деле. А вот чуть повыше мы находим СDS с нужными координатами (обведено зелёным) и видим, что продуктом гена является на самом деле субъединица А ДНК-гиразы, что и хотелось показать. Вопрос о том, почему в npge импортировалась не та аннотация, остаётся открытым.
Проблема 3. В блоке s3x2634 CAI не распознан как старт-кодон в геноме JH1. Это должно насторожить, потому что конец гена совпадает во всех трех последовательностях, и в целом они достаточно схожи друг с другом на всём протяжении гена (и блока тоже, его Identity=98.61%).

Комментарии к работе
Подводя итоги, скажу, что была приятно удивлена функционалом программы npge. В особенности порадовал графический интерфейс — то удобство, которого часто лишены биоинформатические программы.
Задание 2 этого практикума помогло составить общее впечатление о том, что вкладывается в понятие нуклеотидного пангенома, как в общих чертах работает алгоритм его построения, какие параметры существуют для сравнения геномов, по каким блокам выравнивания геномов можно проследить конкретный тип эволюционных изменений. Думаю, было действительно полезно проделать подобный анализ своими руками.
[1] — Wiki