Ресеквенирование. Поиск полиморфизмов у человека
Часть I
Следующие файлы лежат в директории /nfs/srv/databases/ngs/emkeller
- chr4.fasta — человеческая хромосома 4 из сборки генома версии hg19;
- chr4.fastq — файл с одноконцевыми чтениями.
Анализ качества прочтений был проведён при помощи программы FastQC. Для улучшения качества использовался Trimmomatic. Изначально было 5810 чтений с длиной от 43 до 100, после обработки их число уменьшилось до 5715, причём остались только риды с длиной от 50 до 100.
Качество прочтения нуклеотидов
| До чистки Trimmomatic |
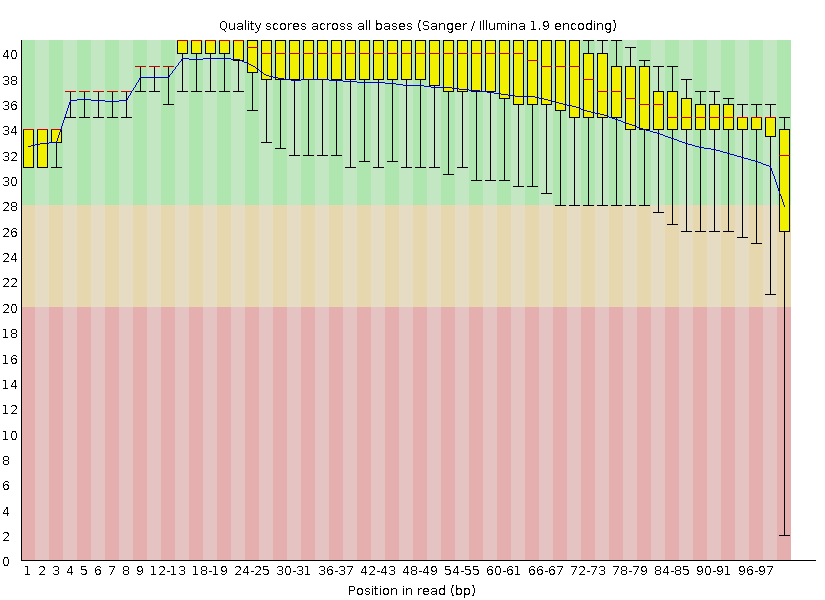 |
| После чистки Trimmomatic |
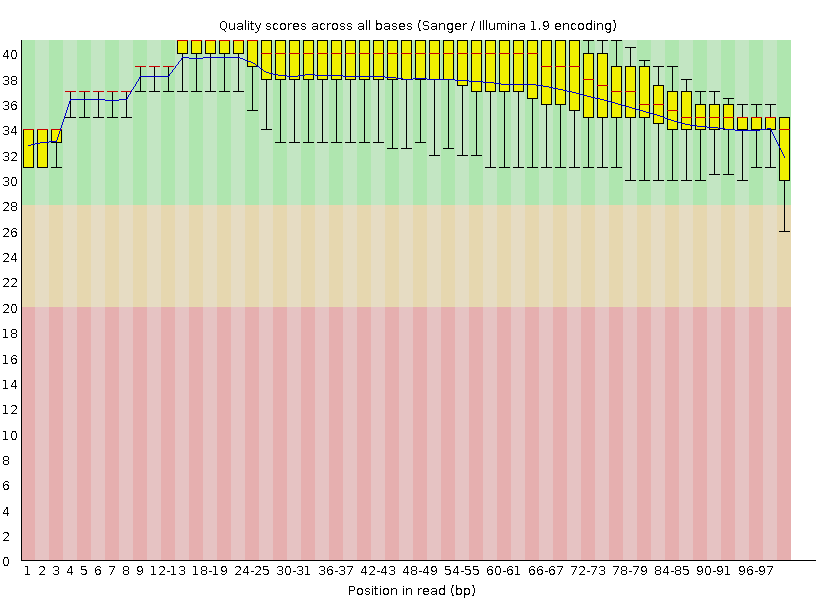 |
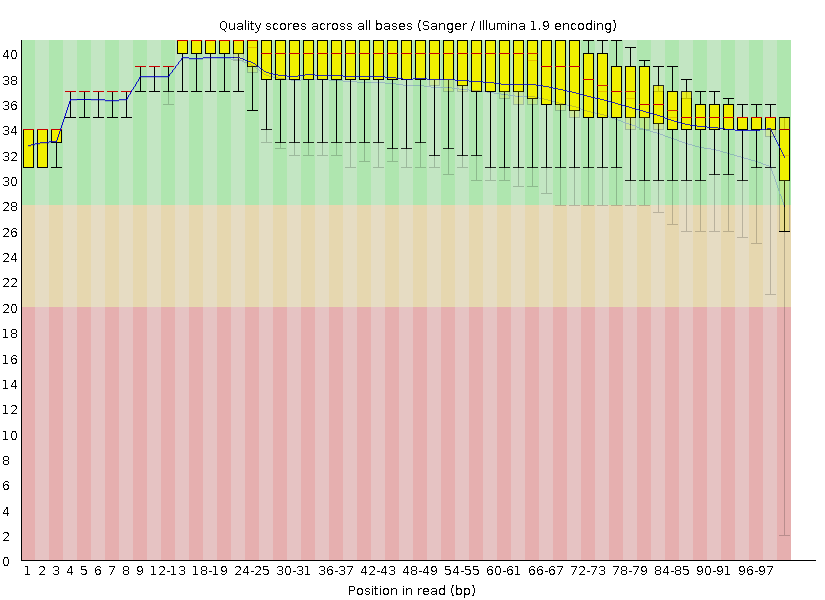
Для удобства два изображения были склеены в одно (Рис. 1). Основное изображение — гистограмма для качества после обработки Trimmomatic, полупрозрачный слой — изначальное состояние.
Качество, очевидно, повысилось, и теперь почти все позиции нуклеотидов прочтены хорошо (попадание в зелёную область гистограммы, quality от 28 и выше). Хорошо видно, что самое мощное повышение качества произошло на конце ридов, что вполне ожидаемо, так как Trimmomatic обрезал концевые нуклеотиды, имевшие качество ниже 20.
Рис. 1. Эффект Trimmomatic
Также можно рассмотреть изображения из выдачи FastQC, на которых показано распределение качества по последовательностям в целом. Здесь важно отметить то, что меняется шкала по горизонтальной оси, на которой отмечается среднее качество последовательности. До обработки шкала начиналась с 2, после же — с 21. Это очень наглядно показывает работу Trimmomatic, ибо, как следует из графиков, он действительно отсекает последовательности с низким (<20) phred-score (качеством).
Распределение качества прочтений
| До чистки Trimmomatic |
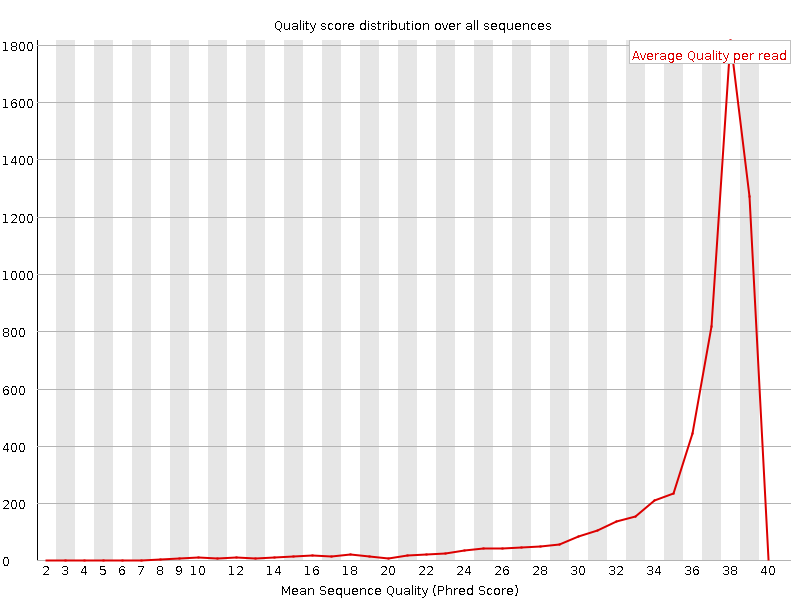 |
| После чистки Trimmomatic |
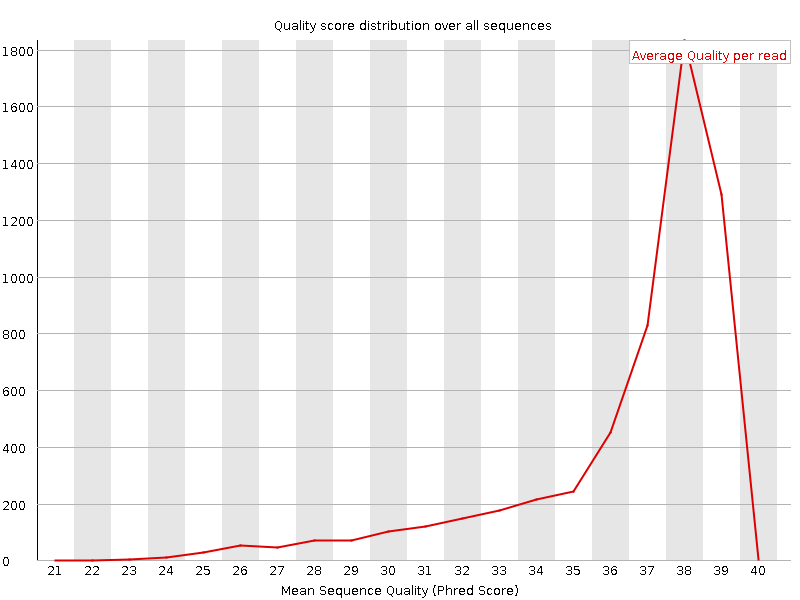 |
Команды. Часть I
| Номер | Команда | Действие |
| 1 | fastqc chr4.fastq | Запуск FastQC для анализа качества чтений. На выходе — [html]-страница с результатами и [zip]-архив. |
| 2 | java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr4.fastq chr4new.fastq TRAILING:20 MINLEN:50 | Запуск Trimmomatic в режиме SE (single-ends) для улучшения качества одноконцевых прочтений. -phred33 |
Часть II
BWA (Burrows-Wheeler aligner) — программа для картирования прочтений. С её помощью производим индексирование референсной последовательности chr4.fasta. Далее будет использоваться в чаcтности алгоритм BWA-MEM, самый точный и быстрый[1] алгоритм для непосредственного построения [sam]-выравнивания прочтений chr4new.fastq с референсом.
Анализ произведенного картирования выполнялся при помощи samtools. В итоге узнаем, что на референсную последовательность картировались все 5715 прочтений, оставшиеся после улучшения качества:
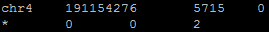
Команды. Часть II
| Номер | Команда | Действие |
| 1 | bwa index chr4.fasta | Индексирование референсной последовательности. |
| 2 | bwa mem chr4.fasta chr4new.fastq > chr4.sam | Картирование чтений на референсную последовательность при помощи алгоритма BWA-MEM. На выходе — выравнивание в формате [sam]. |
| 3 | samtools view chr4.sam -b -o chr4.bam | Перевод [sam]-выравнивания в бинарный [bam] формат. |
| 4 | samtools sort -T /tmp/ -o chr4_sorted.bam chr4.bam | Cортировка [bam]-выравнивания по координате начала прочтения в референсе. Параметр -T нужен для указания пути, по которомусохраняются временные файлы. В итоге имеем файл chr4_sorted.bam |
| 5 | samtools index chr4_sorted.bam | Индекирование отсортированного файла. |
| 6 | samtools idxstats chr4_sorted.bam | Получение информации о количестве картированных прочтений. |
Часть III
Были созданы файл с полиморфизмами (snp.bcf) и файл со списком отличий между референсом и прочтениями (snp.vcf).
Всего найдено 47 полиморфизмов: 42 замены, 4 вставки, 1 делеция. Характеристика трёх полиморфизмов приведена ниже.
Таблица 1. Примеры полифморфизмов
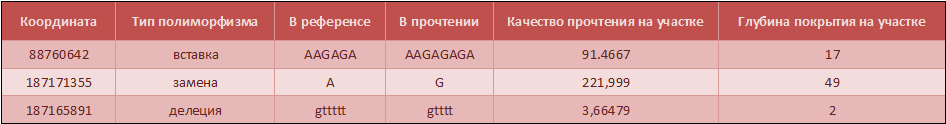
Полученные снипы были проаннотированны по пяти базам данных.
1. Аннотация по базе refgene
В результате было получено три выходных файла: refgeneann.variant_function (информация о всех снипах), refgeneann.exonic_variant_function (информация о снипах, попадающих в экзоны), refgeneann.log (лог работы аннотирующего скрипта).
refgene делит снипы на следующие категории:
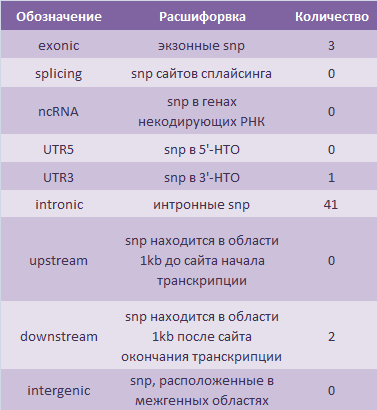
Гены, в которые попали данные SNP: STAP1, MEPE, KLKB1.
Больше всего замен произошло в интронах, что объясняется тем, что эти происшествия не влияют на итоговые продукты генов. В экзонной же области было обнаружено 3 замены, все из них произошли в гене KLKB1 (Kallikrein B1). Из всех замен лишь одна не привела к замене аминокислоты:
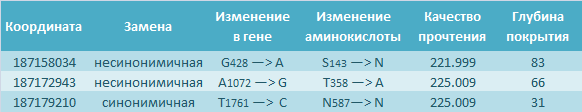
2. Аннотация по базе dbsnp
В результате было получено три выходных файлах: snp138ann.hg19_snp138_dropped (информация о snp, имеющих идентификатор rs в базе данных dbsnp), snp138ann.hg19_snp138_filtered (остальные snp) и snp138ann.log (лог работы скрипта аннотации).
rs-идентификатор имеют 44 snp, 3 — не имеют.
3. Аннотация по базе 1000 genomes
Аннотация по данной БД работает по такому же filter-based-принципу, как и в случае с dbsnp. Выходные файлы аналогичны (_dropped, _filered, .log). Здесь интересно отметить то, что в этом случае неаннотированными стали 7 snp, а не 3, как в предыдущем случае. Причём все snp, неаннотированные в dbsnp так и остались неаннотированными и здесь.
Частота snp варьирует от 0.004 до 0.919. Два из трёх экзонных снипов имеют частоту примерно 0.6, в то время как третий экзонный снип (A1072 —> G) — всего лишь 0.015.
4. Аннотация по базе Gwas
В результате имеем 2 выходных файла: gwasann.hg19_gwasCatalog (снипы, имеющие описанное клиническое значение), gwasann.log (лог работы аннотирующего скрипта).
Ниже можно видеть, что клиническое значение аннотировано в четырёх снипах, причем только один из них является экзонным (выделен рамкой).
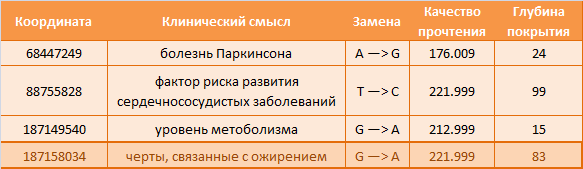
5. Аннотация по базе Clinvar
Здесь мы опять сталкиваемся с filter-based аннотацией и имеем три выходных файла (_dropped, _filered, .log).
В этом случае аннотация была только у одного снипа, причём того же самого, который в прошлом случае был выделен рамкой. Это экзонный снип, имеющий клиническое значение:

Дефицит прекалликреина может приводить к нарушению работы каллекреин-кининовой системы, являющейся ключевой протеолитической системой, участвующей в регуляции широкого спектра физиологических функций организма и развитии многих патологических состояний[2]. В целом же это заболевание является редкой аномалией свертывания крови. Большинство людей с прекалликреиновым дефицитом остаются бессимптомными.
Команды. Часть III
| Номер | Команда | Действие |
| 1 | samtools mpileup -uf chr4.fasta chr4_sorted.bam >snp.bcf | Создание [bcf]-файла с полиморфизмами |
| 2 | bcftools call -cv snp.bcf > snp.vcf | Создание файла со списком отличий между референсом и чтениями в [vcf]-формате |
| 3 | perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp.vcf > snp.avinput | Конвертирование [vcf]-файла в формат [avinput], пригодный для подачи его на вход программе annovar |
| 4 | perl /nfs/srv/databases/annovar/annotate_variation.pl -geneanno -dbtype refGene -buildver hg19 snp.avinput -outfile refgeneann /nfs/srv/databases/annovar/humandb/ | Аннотация полиморфизмов по базе данных refgene |
| 5 | perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype snp138 -buildver hg19 snp.avinput -outfile snp138ann /nfs/srv/databases/annovar/humandb/ | Аннотация полиморфизмов по базе данных dbsnp |
| 6 | perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 snp.avinput -outfile 1000genomesann /nfs/srv/databases/annovar/humandb/ | Аннотация полиморфизмов по базе данных 1000 genomes |
| 7 | perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -dbtype gwasCatalog -buildver hg19 snp.avinput -outfile gwasann /nfs/srv/databases/annovar/humandb/ | Аннотация полиморфизмов по базе данных Gwas |
| 8 | perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19 snp.avinput -outfile clinvarann /nfs/srv/databases/annovar/humandb/ | Аннотация полиморфизмов по базе данных Clinvar |
[1] — Burrows-Wheeler Aligner
[2] — Калликреин-кининовая система