Практикум 10
Определение сайтов связывания ТФ в заданном участке хромосомы человека
Работа велась с файлом chipseq_chunk43.fastq, содержащим риды Illumina, полученные в ходе ChIP-seq эксперимента.
Изначально осуществлялся контроль качества чтений при помощи FastQC, результат чего приведен на Рис. 1a, Рис. 1б.
Рис. 1a. Сводка выдачи FastQC до чистки

Рис. 1б. FastQC: качество по основаниям
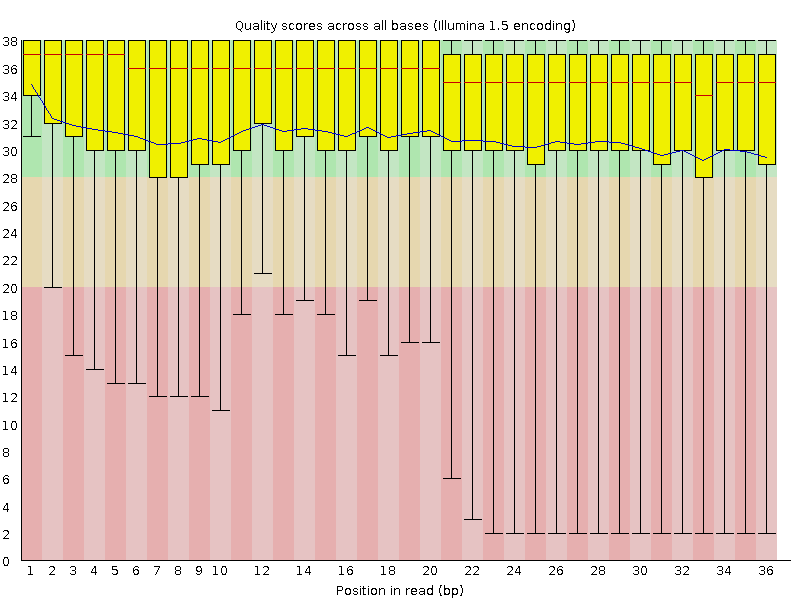
Согласно сводке, файл не содержал последовательностей низкого качества (poor quality). С другой стороны, распределение per base quality оказалось таковым, что наблюдаемые минимумы (концы "усов" на диаграмме) очень сильно заходили в область неприемлемо низкого (<20) качества (красным цветом). Поэтому и было решено провести процедуру очистки, воспользовавшись для этого программой Trimmomatic.
Файл, полученный в результате очистки — chunk43_new.fastqc, содержал 13927 / 17601 последовательностей. Иначе говоря, в ходе работы Trimmomatic отсек порядка 20.8%.
Рис. 2a. Сводка выдачи FastQC после чистки
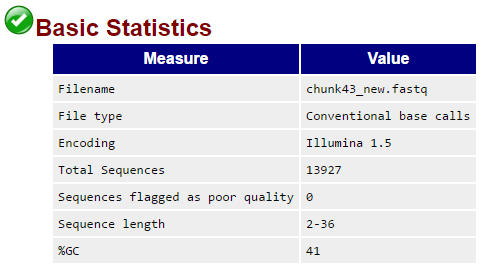
Рис. 2б. FastQC: качество по основаниям
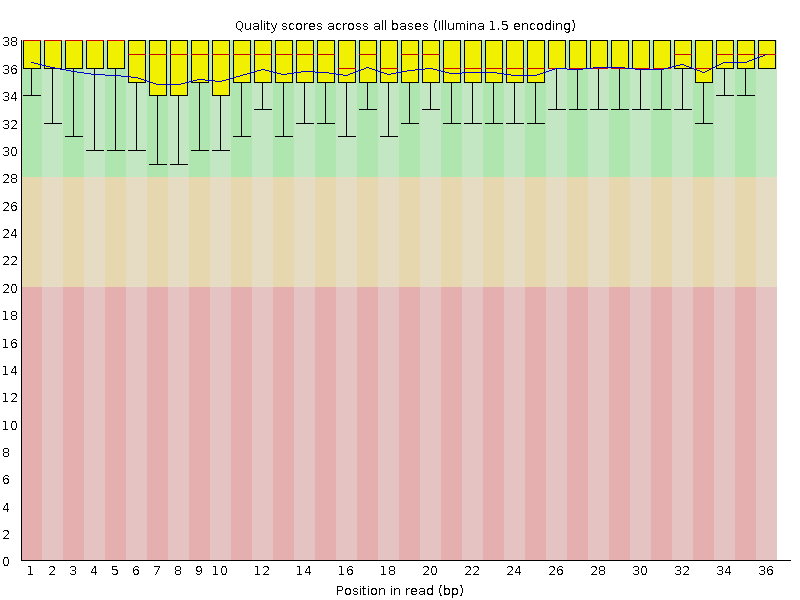
Рис. 2б. радикально отличается от картины, наблюдаемой до процедуры чистки. Теперь качество по каждому нуклеотиду полностью находится в благоприятной (>28) зоне (выделена зеленым). Этому способствовало использование trailing-отсчечения: удаления концевых нуклеотидов, качество которых ниже порогового.
Далее улучшенные риды были откартированы на проиндексированный геном человека при помощи алгоритма BWA-MEM. Анализ картирования (sam-выравнивание: chunk43.sam) проводился при помощи samtools: view для перевода в бинарный формат, sort для сортировки выравнивания по координате начала рида в референсе, index для индексирования, и, наконец,
idxstats для получения информации о количестве прочтений на каждой из хромосом. Информация из файла chunk43_sorted.idxstats приведена на Рис. 3.
Рис. 3. Картирования чтений по хромосомам
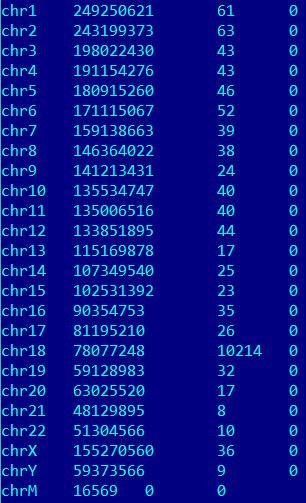
Как видно, риды картировались на все хромосомы, но, исходя из соотношений количеств, будет разумно предположить, что для анализа были предложены прочтения именно с chr18 (10214 ридов).
Процедура поиска пиков осуществлялась с использованием программы MACS2, запуск которой был проведен следующей командой:
macs2 callpeak -t chunk43_sorted.bam --nomodel --outdir callpeak -n chunk43
Параметр outdir задает директорию (callpeak для файлов, в названии которых присутствует имя эксперимента, указываемое в -n (--name). Флаг --nomodel был прописан, поскольку обычный запуск программы
включает в себя построение некоторой модели shifting model, что оказывается невозможным в условиях малого количества пиков (что у нас и наблюдалось).
В результате работы было получено три файла: [narrowPeak], [xls], [bed]. Содержание chunk43_peaks.xls приведено в Таблице 1.
Таблица 1. Информация о пиках из выдачи MACS2
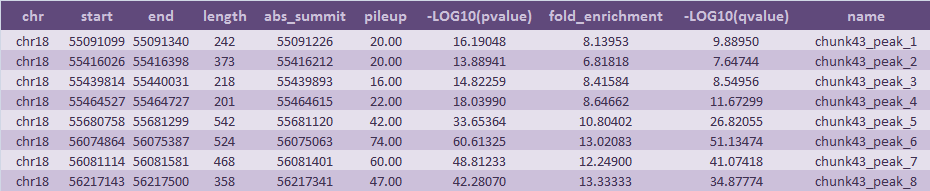
Всего нашлось восемь пиков, принадлежащих 18 хромосоме, ширина которых варьировалась от 201 (peak4) до 542 (peak5). Стоит отметить, что колонки 7 и 9 содержат отрицательные десятичные логарифмы p-value и q-value, а значит трактовать их надо не так, как обычные значения. Здесь работает принцип "чем больше, тем лучше", поскольку при повышении этих значений p-value и q-value падают. По ним можно судить о достоверности пиков, и, согласно этому критерию, все находки оказались достаточно надежными. В частности, самый надежный пик (peak6) имеет показатели:
-log10(p-value): 60.61 и -log10(q-value): 51.13Наихудшие же показатели у peak2:
-log10(p-value): 13.89 и -log10(q-value): 7.65Но тем не менее очевидно, что и этот пик можно назвать достоверным с большой долей уверенности несмотря на то, что на фоне остальных он вышел хуже всего.
Визуализация пиков проводилась в геномном браузере UCSC Genome Browser[1]. Для того чтобы использовать файл [narrowPeaks] в качестве трэк-файла, в него добавились следующие строки:
track type=narrowPeak visibility=3 db=hg19 name="my_peaks" description="Peaks from chunk 43" browser position chr18:55075850-56230000
На Рис. 4 приведен соответствующий скриншот из браузера.
Рис. 4. Визуализация пиков в UCSC Genome Browser
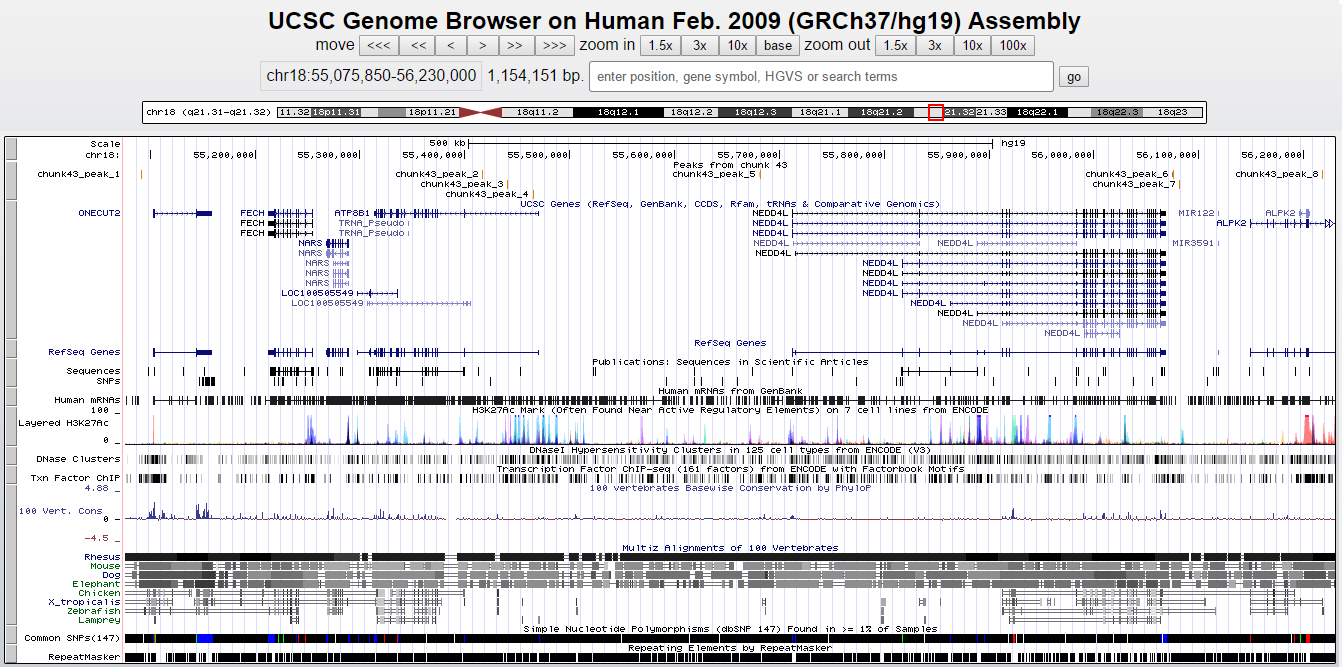
Для более детального рассмотрения был взят peak6 как наиболее надежный (Рис. 5), но ни с какими генами он, как видно, не пересекается.
Рис. 5. Детальное изображение peak6
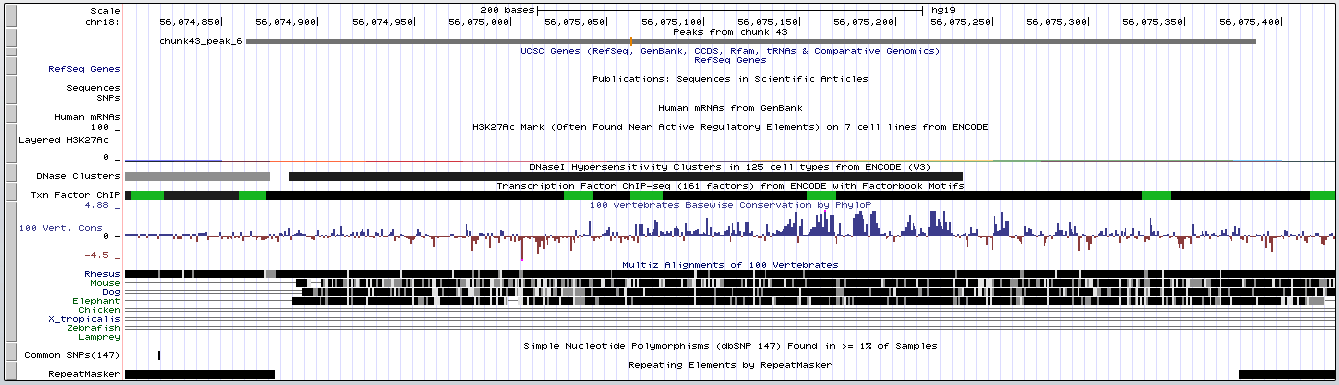
Параметры peak6:
- координаты в геноме:
56074864:56075387; - ширина пика: 524 (самый широкий);
- расстояние от начала до вершины: 199
- расположение вершины в пике: 199:325
- достоверность: -log10(p-value): 60.61 и -log10(q-value): 51.13 (самый достоверный);
Рассматривался также и peak4, скриншот из геномного браузера с приближением которого приведен на Рис. 6.
Рис. 6. Детальное изображение peak4
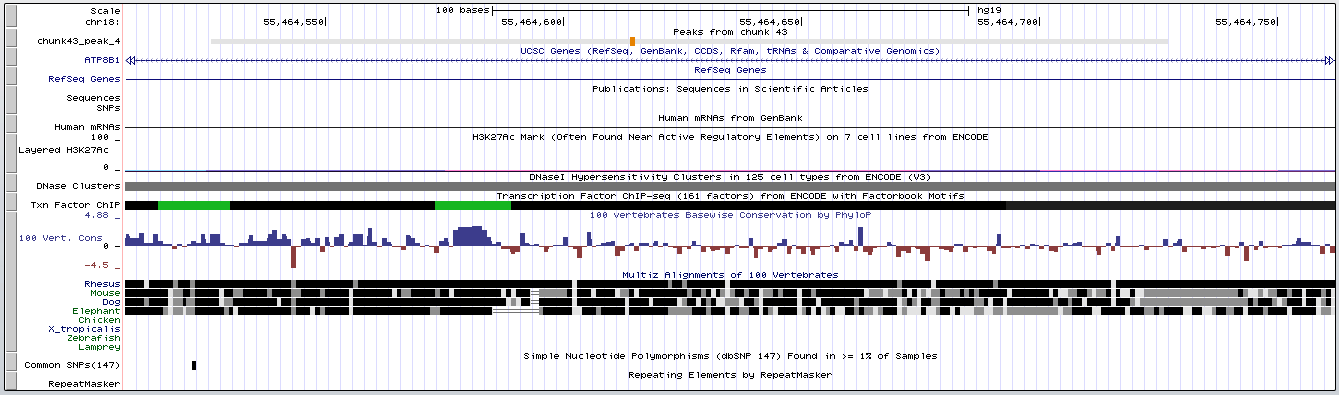
Параметры peak6:
- координаты в геноме:
55464527:55464727; - ширина пика: 201 (самый узкий);
- расстояние от начала до вершины: 88
- расположение вершины в пике: 88:113
- достоверность: -log10(p-value): 18.04 и -log10(q-value): 11.67 (достоверен);
В отличие от предыдущего пика, peak4 пересекается с некодирующей частью гена ATP8B1, который кодирует АТФазу p-типа, выступающую в роли аминофосфолипидного транспортера. Мутации в этом гене могут приводить к развитию внутрипеченочных холестазов: доброкачественного рецидивирующего или прогрессирующего семейного. Механизмы этого процесса на данный момент неизвестны[2].
Файл [narrowPeaks] был переведен в формат [fasta]:
/srv/databases/ngs/tools/seqtk/seqtk subseq /srv/databases/ngs/hg19/GRCh37.p13.genome.fa [].narrowPeak > [].fa
Далее строилось выравнивание последовательностей, результат приведен на Рис. 7.
Рис. 7. Выравнивание последовательностей пиков

Выравнивание получилось низкого качества. В частности же малый процент консервативных колонок и огромное количество гэпов не позволяют выделить консенсус длиной 4-8 нуклеотидов.
[1] UCSC Genome Browser
[3] Progressive familial intrahepatic cholestasis