Работа с KEGG ORTHOLOGY
В заданиях данного практикума проводилась работа с базой данных KEGG.
Выбор пары ортологических рядов
Для исследования был выбран метаболизм пируватов (англ. pyruvate metabolism), представленный на Рис. 1. На нём же зелёным цветом выделена реакция с EC 1.2.1.10.
Рис. 1. Схема метаболизма пируватов из KEGG
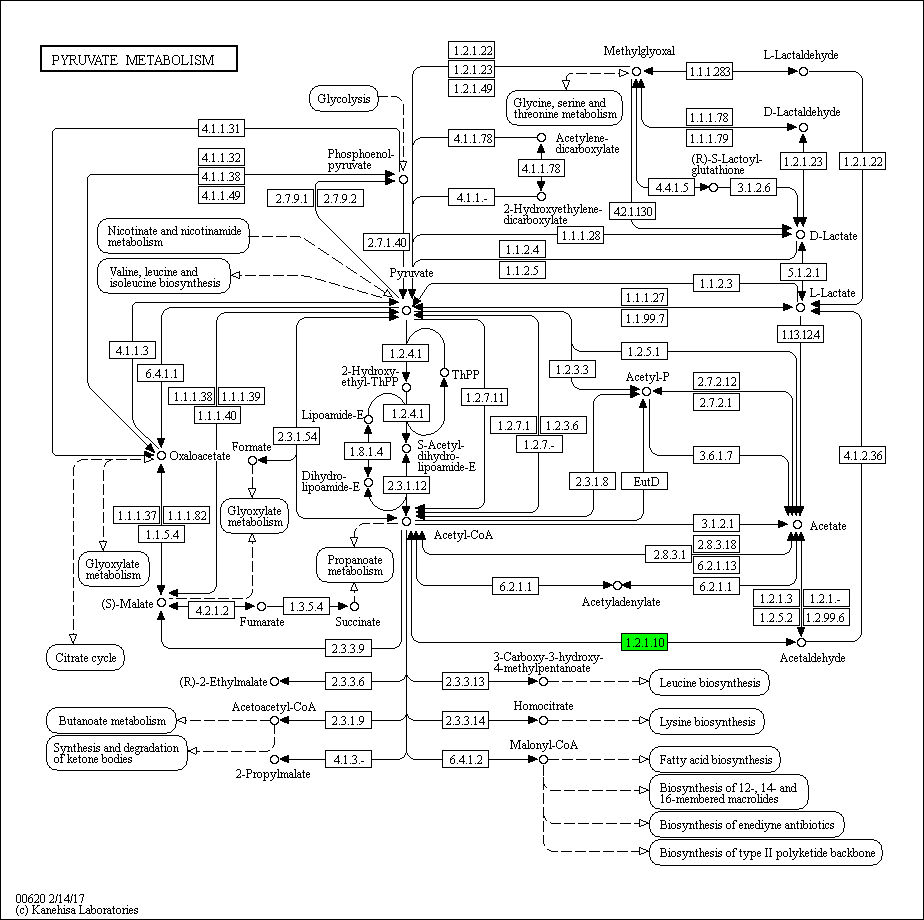
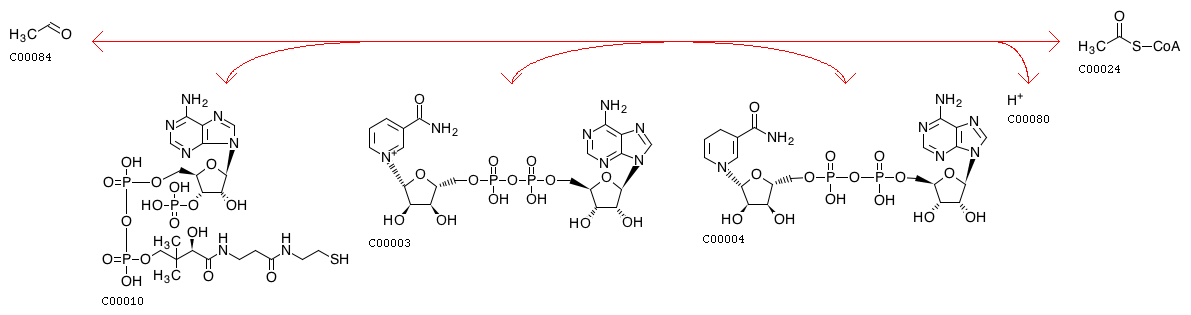
Схема указанной реакции (идентификатор: R00228) представлена на Рис. 2. В ходе неё ацетальдегид реагирует с коэнзимом-А, образуя ацетил-коэнзим А, причем попутно происходит восстановление NAD+ до NADH. Ферменты, катализирующие данную реакцию, относятся к:
>оксидоредуктазам
>действующим на альдегидную или оксо-группу донора
>имеющим NAD+/NADP+ в качестве акцептора
Рис. 2. Схема реакции R00228
Всего в KEGG общее число ортологичных рядов составило пять штук. Однако из них были отобраны два, — K18366 и K04021, — исходя из соображений удобства работы, что оценивалось по количеству последовательностей в ряду. Стоит отметить, что среди неподходящих рядов был как и очень небольшой ряд с 40 последовательностями, так и длинные ряды в 456 и 777 последовательностей. Выбранные же для дальнейшей работы ряды содержали 100 и 101 последовательность соответственно, то есть, подходили идеально: можно рассчитывать на то, что не возникнет проблем с длительностью построения выравнивания и дерева.
Получение совместного множественного выравнивания
Для каждого из выбранных ортологических рядов средствами базы данных Uniprot (Retrieve/ID Mapping) были получены последовательности всех входящих в них белков (K18366.fasta, K04021.fasta).
Затем для модификации названий (с целью учета ортологического ряда) был написан специальный скрипт на Python, и в итоге был получен файл с последовательностями всех белков обоих рядов с наглядными названиями (seqs.fasta).
В программе Jalview при помощи сервиса Muscle было построено множественное выравнивание. Доступен [fasta]-файл с выравниванием, а также итоговый проект [jvp] (окно: align_seqs.fasta).
Проверка выравнивания
Некоторые из белков действительно очень сильно выбивались из выравнивания: какие-то из них были просто плохо выровнены на всем протяжении последовательности. Для трех таких последовательностей было построено выравнивание в отдельном окне out1.fasta, по которому становится очевидно (Рис. 3), что данные последовательности принадлежат отдельному ортологическому ряду, и, следовательно, должны быть удалены из общего выравнивания.
Рис. 3. Отдельный ортологический ряд, подлежащий удалению

Другие же последовательности, обаладая малой длиной (в сравнении с типичной длиной белка в K18366: ~300, в K04021: ~470), имели большое количество гэпов в областях консервативных (для множества остальных последовательностей) блоков. Такие последовательности тоже были удалены из общего выравнивания.
Гомологичность белков в выравнивании
После описанного избавления от ненужных последовательностей было получено выравнивание в окне check_align_seqs.fasta. Оно приведено на Рис. 4.
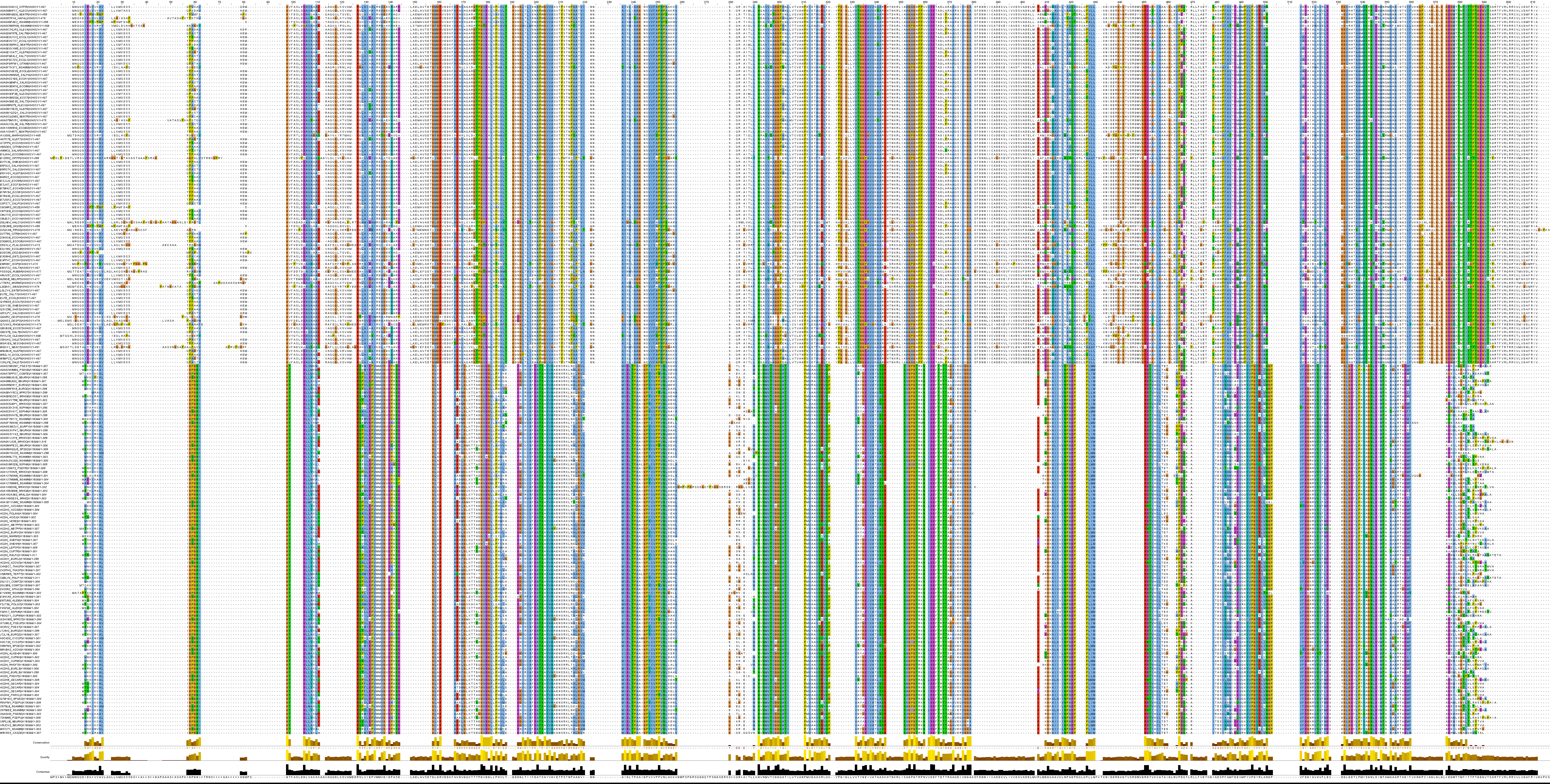
К сожалению, изначально я допустила ошибку, посчитав это выравнивание хорошим в том числе и на основании того, что прослеживалась консервативность редких аминокислот. Это не так, поскольку для увеличения общего веса выравнивания алгоритму при любых условиях "выгоднее" выровнять эти дорогие по весу аминокислоты. Поэтому, наблюдая колонки из редких аминокислот, нельзя терять бдительности. В целом же выравние действительно плохого качества. Поэтому построение по нему дерева являлось непростительной ошибкой, ведущей к ничем необоснованным выводам.