Практикум 11. Молекулярная динамика биологических молекул в GROMACS. Моделирование самосборки липидного бислоя.
Целью работы было моделирование самосборки липидного бислоя. Работа выполнялась частично на kodomo, частично же — на суперкомпьютере Ломоносов-2.
Часть файлов была предоставлена изначально:
- файл дополнительной топологии для липида DPPC, dppc.itp.
- файл параметров для липидов, lipid.itp
- файл с координатами одного липида, dppc.gro
- файл-заготовка топологии системы, b.top
- файл праметров для минимизации энергии, em.mdp
- файл пaраметров для "утряски" воды, pr.mdp
- файл пaраметров для молекулярной динамики, md.mdp
На основе файла с координатами одного липида была создана ячейка, содержащая 43=64 липида:
gmx genconf -f dppc.gro -o b_64.gro -nbox 4 4 4
Далее было проведено преобразование *.gro файлов в *.pdb:
gmx editconf -f b_64.gro -o b_64.pdb
gmx editconf -f dppc.gro -o dppc.pdb
Полученные структуры были просмотрены в PyMol (см. Рис. 1 и Рис. 2).
Рис. 1. Структура липида DPPC
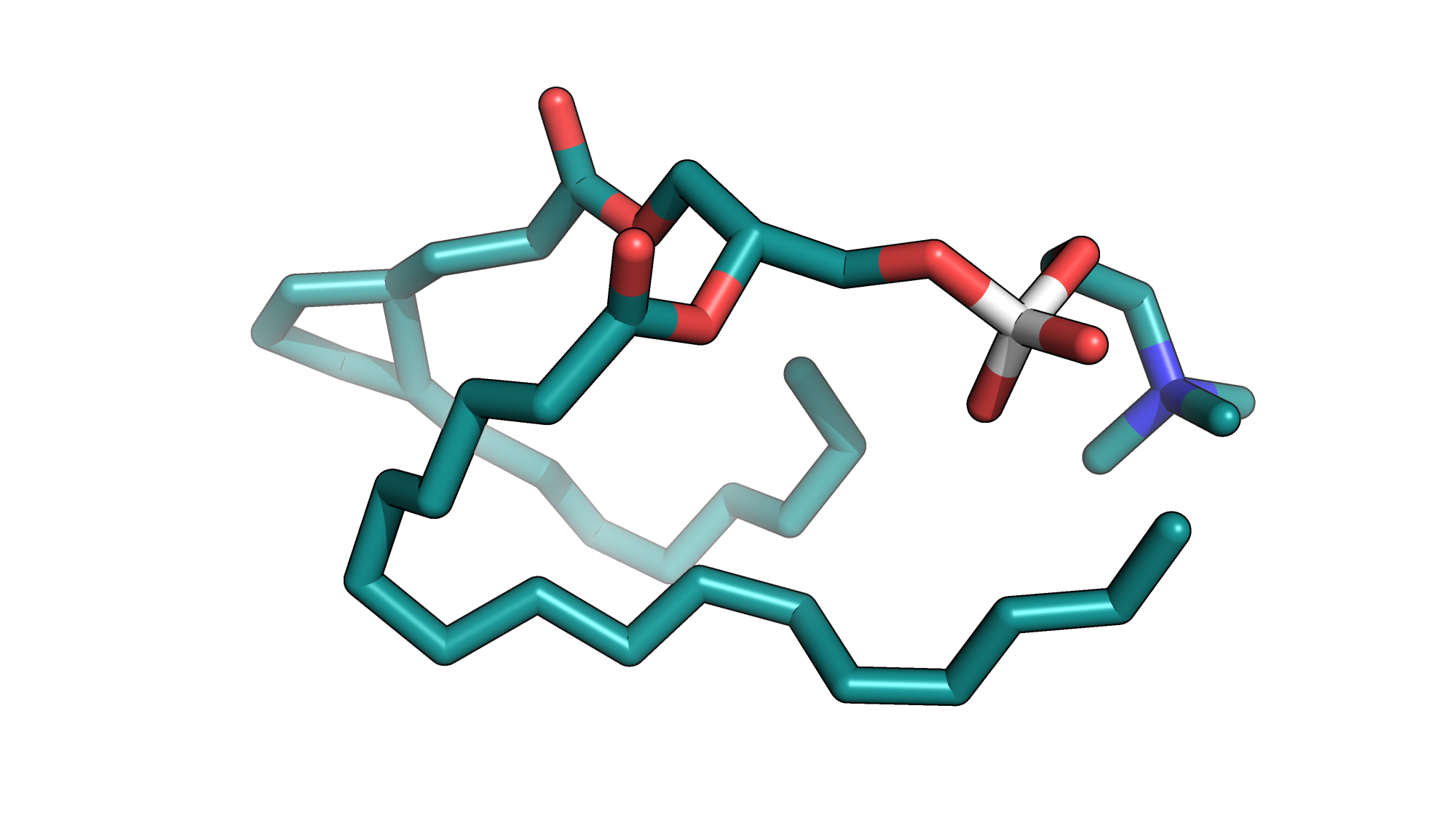
Рис. 2. Структура ячейки c 64 молекулами DPPC

В ячейке был сделан отступ:
gmx editconf -f b_64.gro -o b_ec -d 0.5
Была проведена оптимизация геометрии системы с целью избавления от некорректных контактов:
gmx grompp -f em -c b_ec -p b -o b_em -maxwarn 2
gmx mdrun -deffnm b_em -v
В ходе оптимизации геометрии произошло изменение максимальной силы: от начального значения 2.17409e+07 до конечного 2.36037e+03.
В ячейку была добавлена single-point charge (spc) вода:
gmx solvate -cp b_em -p b -cs spc216 -o b_s
Для утряски воды во избежание взрыва системы провели следующие операции:
gmx grompp -f em -c b_s -p b -o b_empr -maxwarn 1
gmx mdrun -deffnm b_empr -v
gmx grompp -f pr -c b_empr -p b -o b_pr -maxwarn 1
gmx mdrun -deffnm b_pr -v
Далее вновь было проведено преобразование *.gro файлов в *.pdb:
gmx editconf -f b_s.gro -o b_s.pdb
gmx editconf -f b_pr.gro -o b_pr.pdb
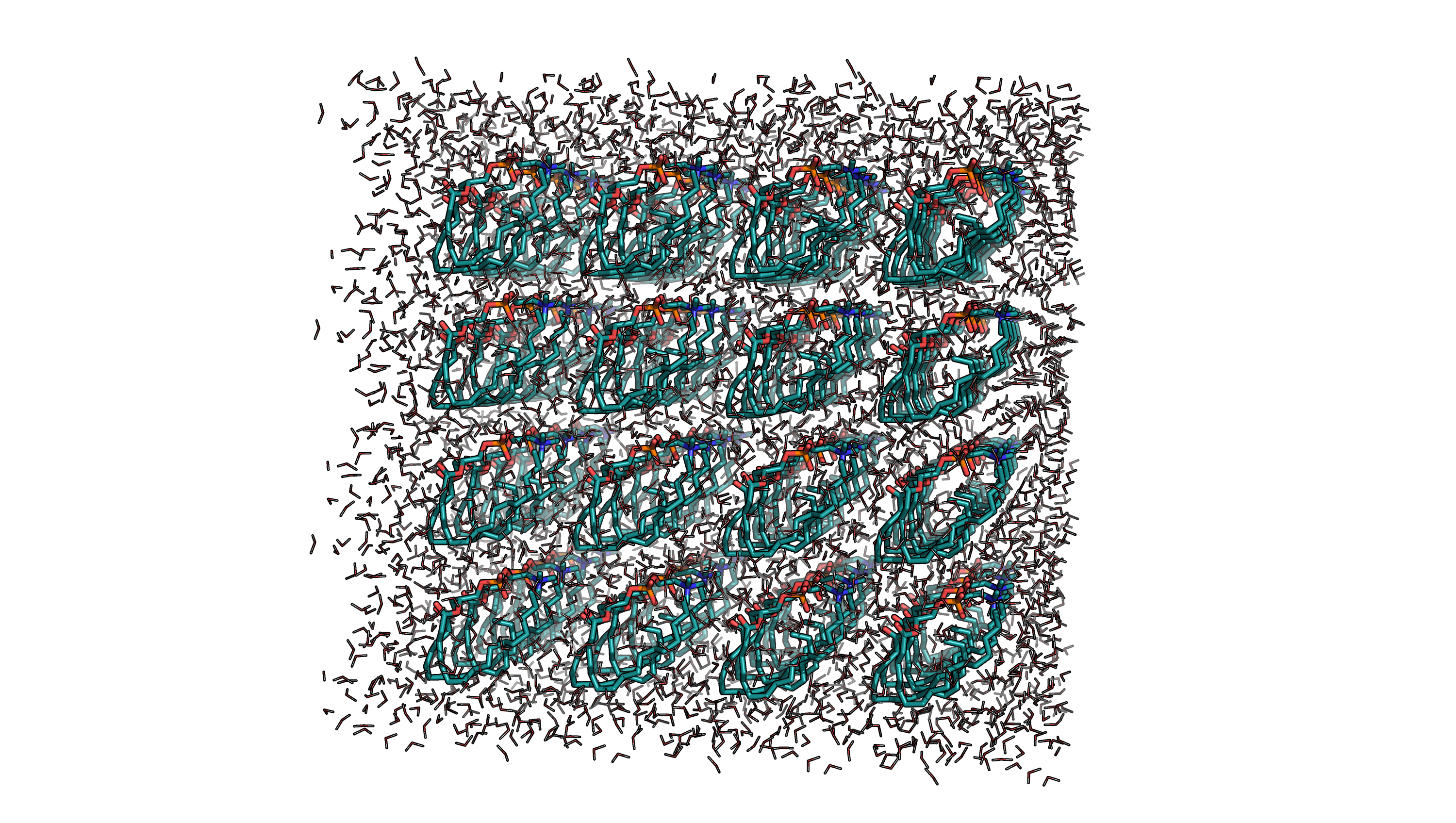
Полученные структуры были просмотрены в PyMol (см. Рис. 3 и Рис. 4).
Рис. 3. До утряски воды |
Рис. 4. После утряски воды |
 |
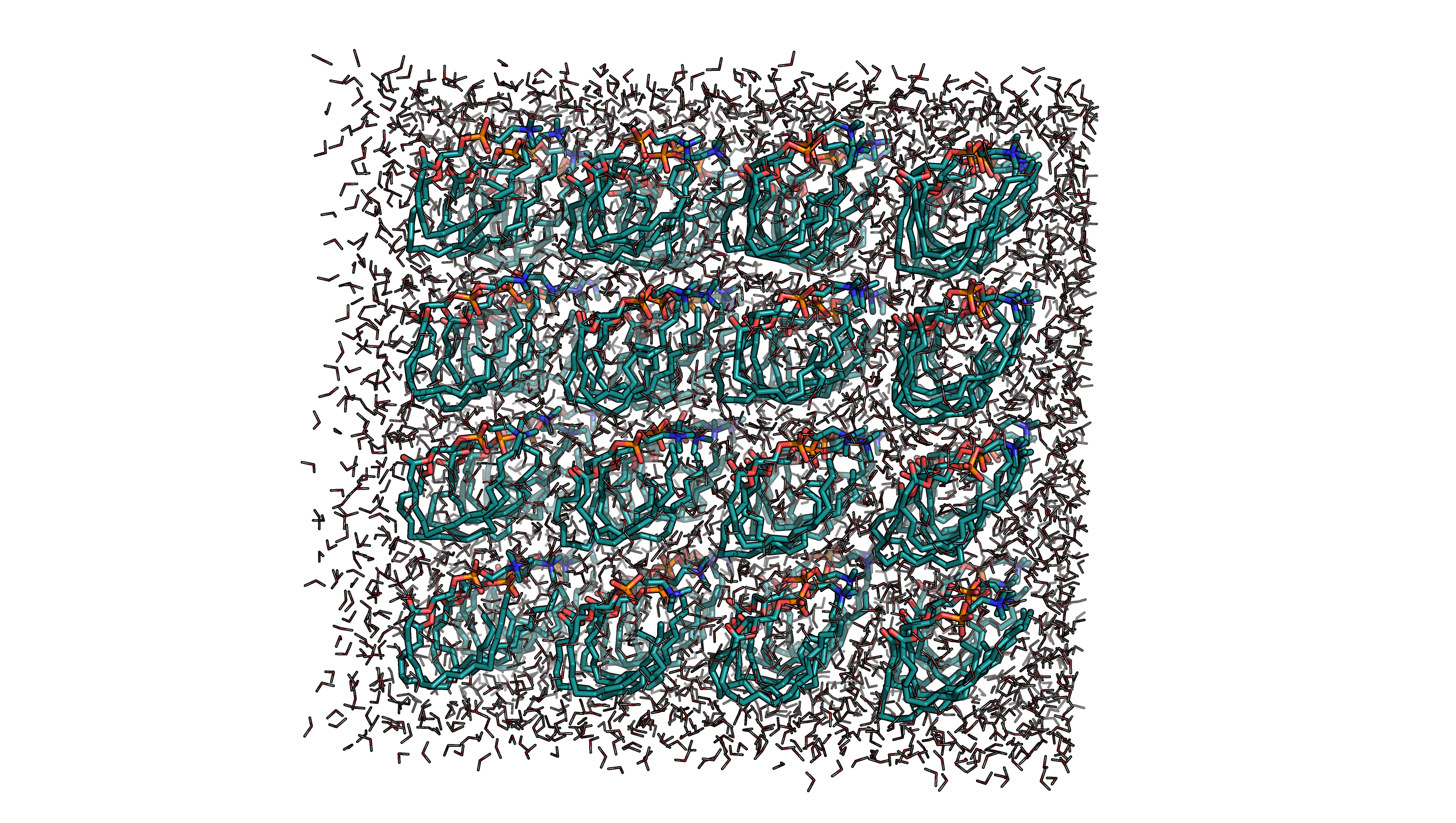 |
Видно, как вода распределилась более естественно по ячейке.
Далее на суперкомпьютере Ломоносов-2 было запущено тестовое моделирование:
Подключаем GROMACS
module load gromacs/2020.3-gcc-gpu openmpi/1.8.4-gcc
Копируем в свою директорию необходимые файлы
cp /home/golovin/progs/gromacs-2016.3/share/top/residuetypes.dat .
cp -r /home/fbbstudent/_scratch/fbb/gmx.ff .
Ставим задачу на расчет на 5 минут:
gmx grompp -f md -c b_pr -p b -o b_md -maxwarn 1
sbatch -N1 --ntasks-per-node=1 -e error.log -o output.log -t 5 -p test ompi /opt/ccoe/gromacs-2020.3-gcc-cuda/bin/gmx mdrun -deffnm b_md -v
Тестовый запуск прошел успешно.