Практикум 11
| команда | зачем |
|---|---|
| cp Human/chr17.fasta esiling/chr17.fasta | Копирование необходимых файлов в свою директорию |
| hisat2-build chr17.fasta chr17 | Индексация референса |
| cp ../Human/reads/chr8.fastq chr17.fastq | Копирование |
| fastqc chr17.fastq | Анализ программой FastQC. Потом смотреть на созданный html файл |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr17.fastq chr17_trimmed.fastq TRAILING:20 MINLEN:50 | Очистка при помощи Trimmomatic. Адаптеров уже не было изначально, чтения одноконцевые. Необходимо только обрезать с конца нуклеотды с качество ниже 20, и убрать все последовательности длиной менее 50 нк |
| hisat2 -x chr17 -U chr17_trimmed.fastq -S chr17_aligntoref.sam --no-softclip --no-spliced-alignment | Картирование чтений из fastq файла на проиндексированную последовательность; --nosoftclip - выравнивание должно быть по всему риду; --no-spliced-alignment - выравнивание не должно допускать гэпов и инделей в риде |
| samtools view -b chr17_aligntoref.sam -o chr17_aligntoref.bam | Перевод в бинарный bam формат |
| samtools sort chr17_aligntoref.bam chr17_altoref_sorted | Сортировка выравниваний по координате в референсе |
| samtools index chr17_altoref_sorted.bam | Индексирование |
| samtools mpileup -u -f chr17.fasta -o chr17_polymorph.bcf chr17_altoref_sorted.bam | Создание файла с полиморфизмами |
| bcftools call -cv -o chr17_polymorph.vcf chr17_polymorph.bcf | Изменение формата, -v - output variant sites only - то есть показать только изменившиеся позиции |
| vcftools --vcf chr17_polymorph.vcf --remove-indels --recode --out chr17_polymorph_noindel | Удалить из vcf файла индели (удалилось 4 инделя) |
| convert2annovar.pl -format vcf4 chr17_polymorph_noindel.recode.vcf -outfile chr17_polymorph.avinput | Конвертация в формат, который распознает annovar |
| annotate_variation.pl -out ann_chr17_refgene -build hg19 -dbtype refGene chr17_polymorph.avinput /nfs/srv/databases/annovar/humandb.old/ | Аннотация по refgene, на генной разметке |
| annotate_variation.pl -filter -out ann_chr17_dbsnp -build hg19 -dbtype snp138 chr17_polymorph.avinput /nfs/srv/databases/annovar/humandb.old/ | Аннотацтия по dbsnp, основана на фильтрации |
| annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out ann_chr17_1000g chr17_polymorph.avinput /nfs/srv/databases/annovar/humandb.old/ | Аннотацтия по 1000 genomes, основана на фильтрации |
| annotate_variation.pl -regionanno -build hg19 -out ann_chr17_gwas -dbtype gwasCatalog chr17_polymorph.avinput /nfs/srv/databases/annovar/humandb.old/ | Аннотацтия по GWAS, основана на разметке других регионов генома |
| annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19 -out ann_chr17_clinvar chr17_polymorph.avinput /nfs/srv/databases/annovar/humandb.old/ | Аннотацтия по Clinvar, основана на фильтрации |
1. Анализ качества чтений
скачать исходный .fastq файл хромосомы
скачать триммированный .fastq файл
Подготовка: индексирование хромосомы командой hisat2-build chr17.fasta indexed.
Я пользуюсь OSX, и мне было интересно скачать FastQC как программу и работать не в командной строке, а с интерфейсом FastQC. Я скачала ее с официального сайта (чертыхаясь и немного мучаясь). Зато удобно.
Запуск программы для необрезанной последовательности показал 11046 прочтений, из них с низким качеством ни одного, но давайте посмотрим на картинку:
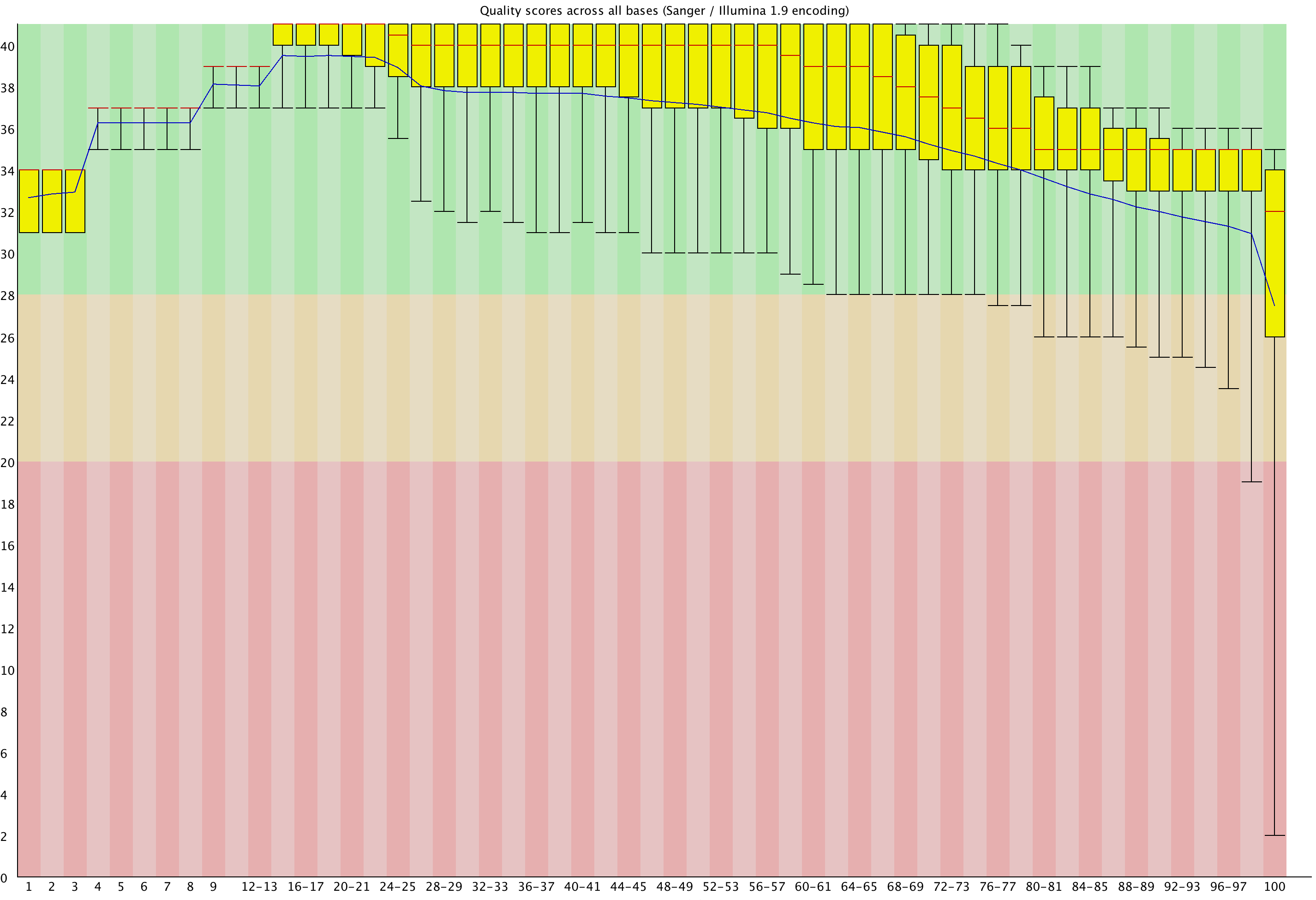
Это можно исправить триммированием последовательности:
jar SE -phred33 chr17.fastq chr17.trimmed.fastq TRAILING:20 MINLEN:50
Картинка после триммирования:
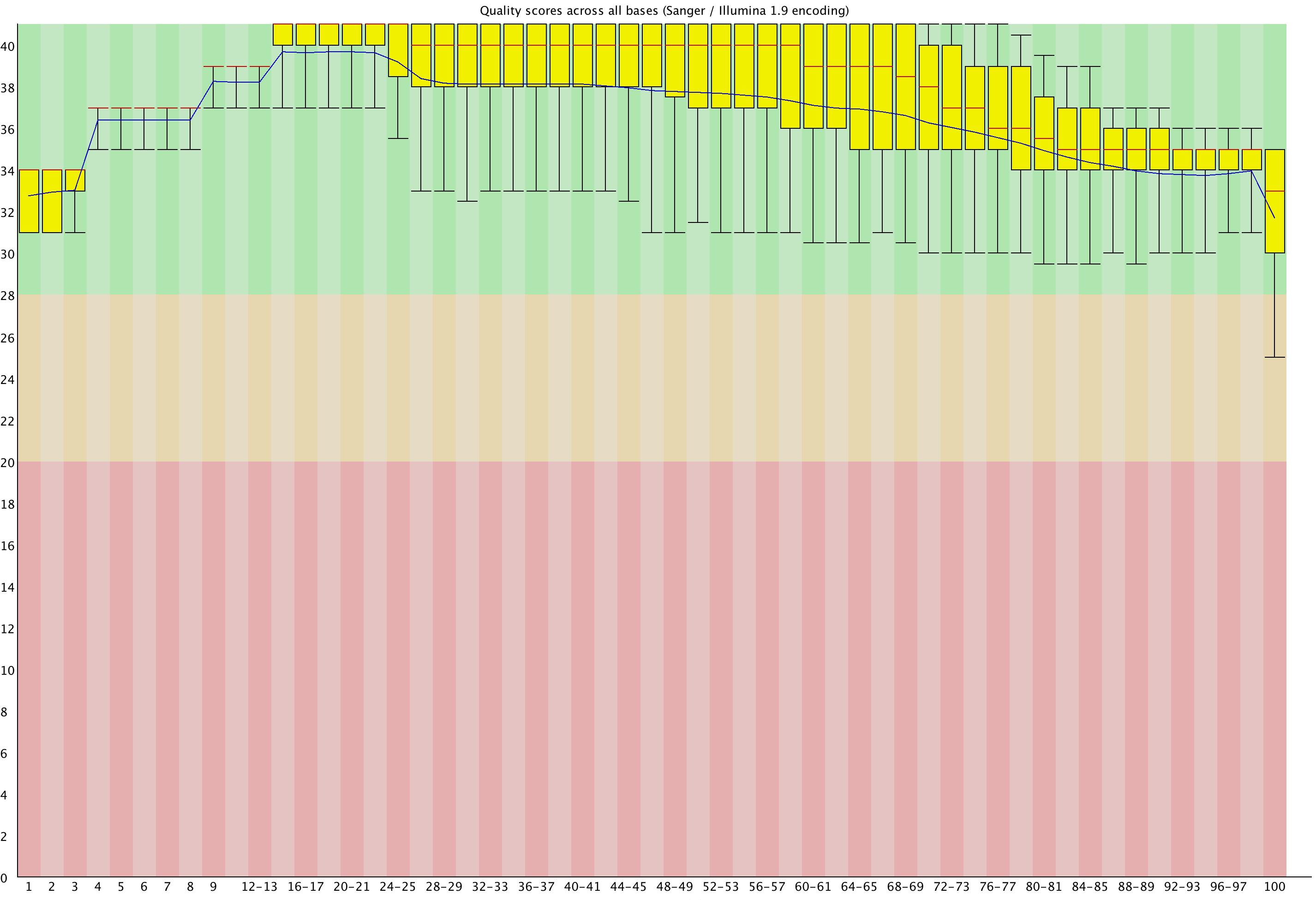
Отдельно хочется отметить, как триммирование повлияло на распределение G/C содержания в последовательности. До триммирования FastQC выдавал "fail":
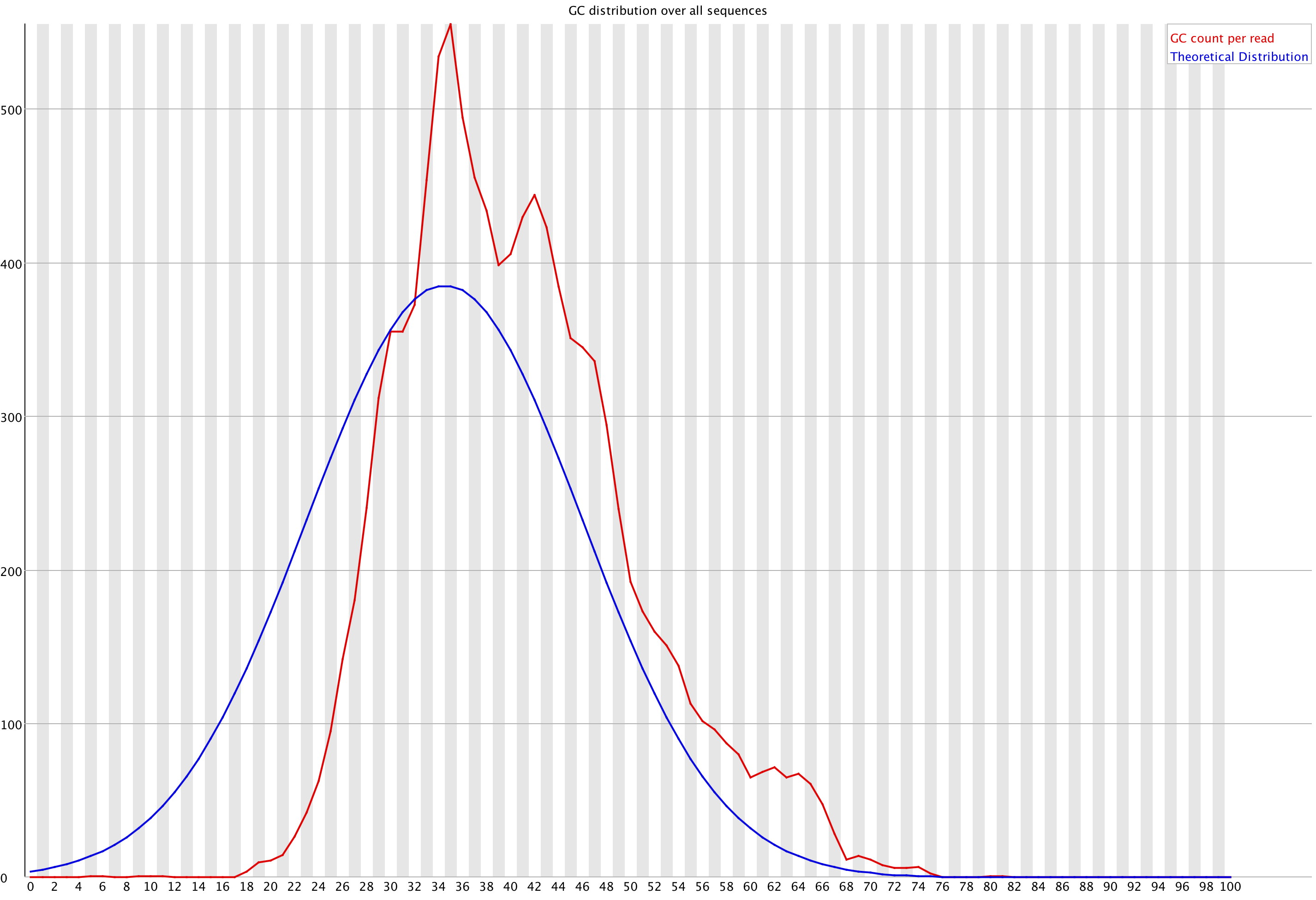
Как отмечали в своих практикумах мои коллеги, зачастую триммирование позволяет получить картинку распределения GC-содержания, более похожего на нормальное. К сожалению (и пока не могу, наверное, объяснить, из-за чего) с моей хромосомой после обрезки в плане GC ничего существенно не изменилось:
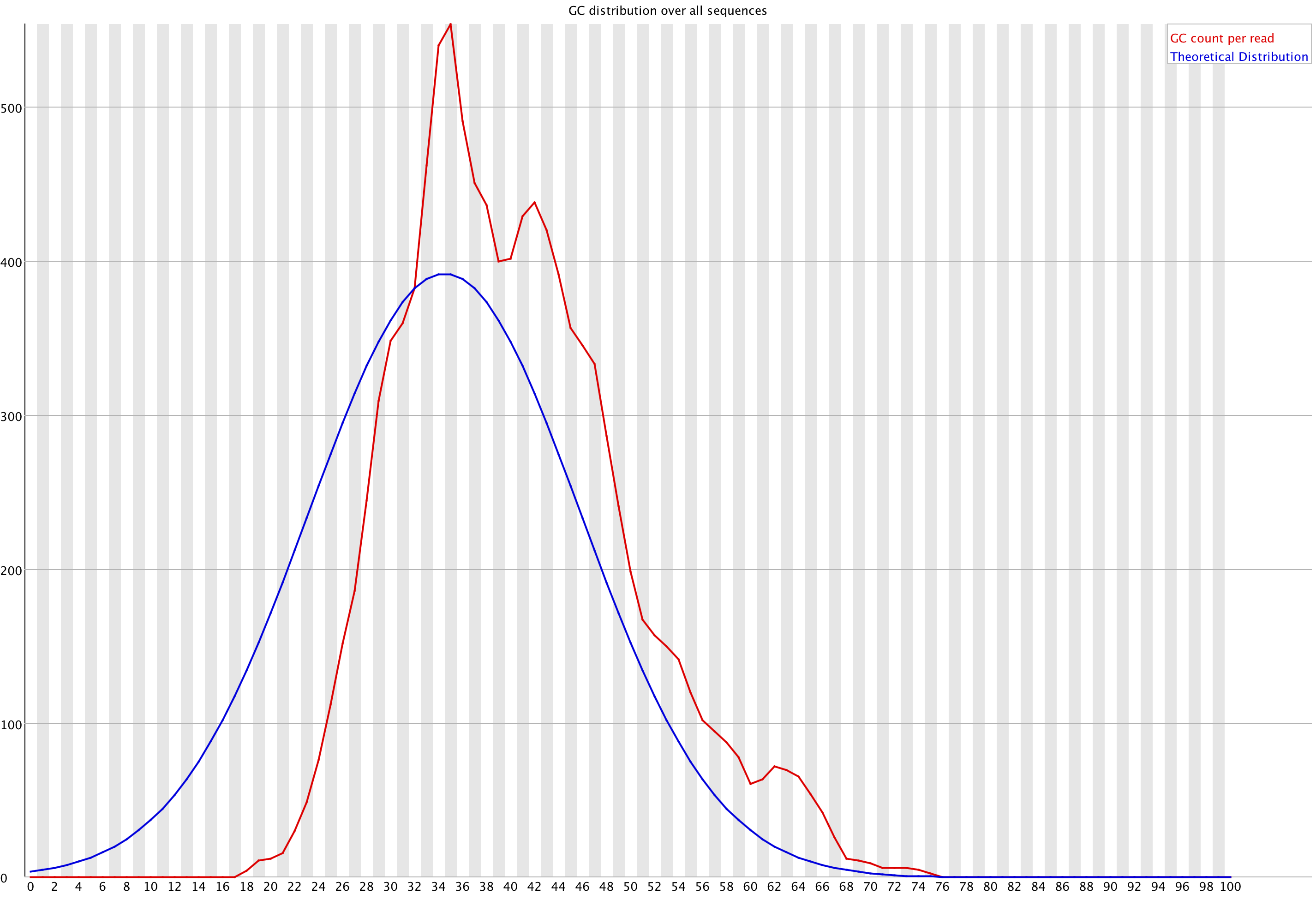
Картирование чтений
Результат после работы hisat2:
10868 reads; of these:
10868 (100.00%) were unpaired; of these:
34 (0.31%) aligned 0 times
7247 (66.68%) aligned exactly 1 time
3587 (33.01%) aligned >1 times
вывод - качество картирования хорошее
Команда samtools flagstat показывает статистику:
14424 + 0 mapped (99.76%:-nan%)
"After filtering, kept 54 out of a possible 58 Sites" - после удаления инделей из .vcf файла (*)
Описание трех полиморфизмов из .vcf файла
| координата | тип полиморфизма | референс | чтения | глубина покрытия | качество чтения |
|---|---|---|---|---|---|
| 44788253 | замена | G | A | 72 | 221.999 |
| 79534241 | делеция | ATCTTTCT | ATCT | 4 | 13.657 |
| 44832598 | замена | С | Т | 32 | 225.009 |
Анализ однонуклеотидных полиморфизмов (SNP)
Вышло 38 snp и 4 инделя (см *, на деле строчек 54, но в них говорится о 38 snp, что-то растянулось на 2 строчки)
Качество:
медиана 12,49995
среднее 69,08629
максимум 225,009
минимум 3.01497
| категория | количество SNP в группе |
|---|---|
| UTR3 | 5 |
| intronic | 30 |
| exonic | 3 |
| строка | ген | замена |
|---|---|---|
| 22 | CD79B:NM_000626:exon3 | synonymous SNV A -> G |
| 49 | NPLOC4:NM_017921:exon3 | synonymous SNV G -> A |
| 53 | NPLOC4:NM_017921:exon2 | synonymous SNV C -> T |
Клиническая аннотация
Clinvar, к сожалению, ничего не нашел. Выдача GWAS:
gwasCatalog Name=Ovarian cancer in BRCA1 mutation carriers,Parkinson's disease chr17 44788310 44788310 G A hom 221.999 . gwasCatalog Name=Height chr17 62007498 62007498 A G hom 221.999 . gwasCatalog Name=Eye color traits chr17 79596811 79596811 C T hom 221.999 .