Практикум 1. Филогенетическое дерево
Отобранные бактерии
| # | название | мнемоника |
|---|---|---|
| 1 | Bacillus anthracis | BACAN |
| 2 | Clostridium botulinum | CLOBA |
| 3 | Enterococcus faecalis | ENTFA |
| 4 | Geobacillus kaustophilus | GEOKA |
| 5 | Lactobacillus acidophilus | LACAC |
| 6 | Listeria monocytogenes serovar 1/2a | LISMO |
| 7 | Moorella thermoacetica | MOOTA |
| 8 | Staphylococcus aureus | STAAR |
| 9 | Streptococcus pyogenes serotype M1 | STRP1 |
| 10 | Streptococcus pneumoniae serotype 4 | STRPN |
Скобочная формула дерева
((MOOTA, CLOBA), ((((GEOKA, BACAN), (LISMO)), (STAAR)), ((ENTFA), ((LACAC), (STRPN, STRP1)))));
Изображение дерева
Изображение получено при визуализации скобочной формулы дерева в программе MEGA X.
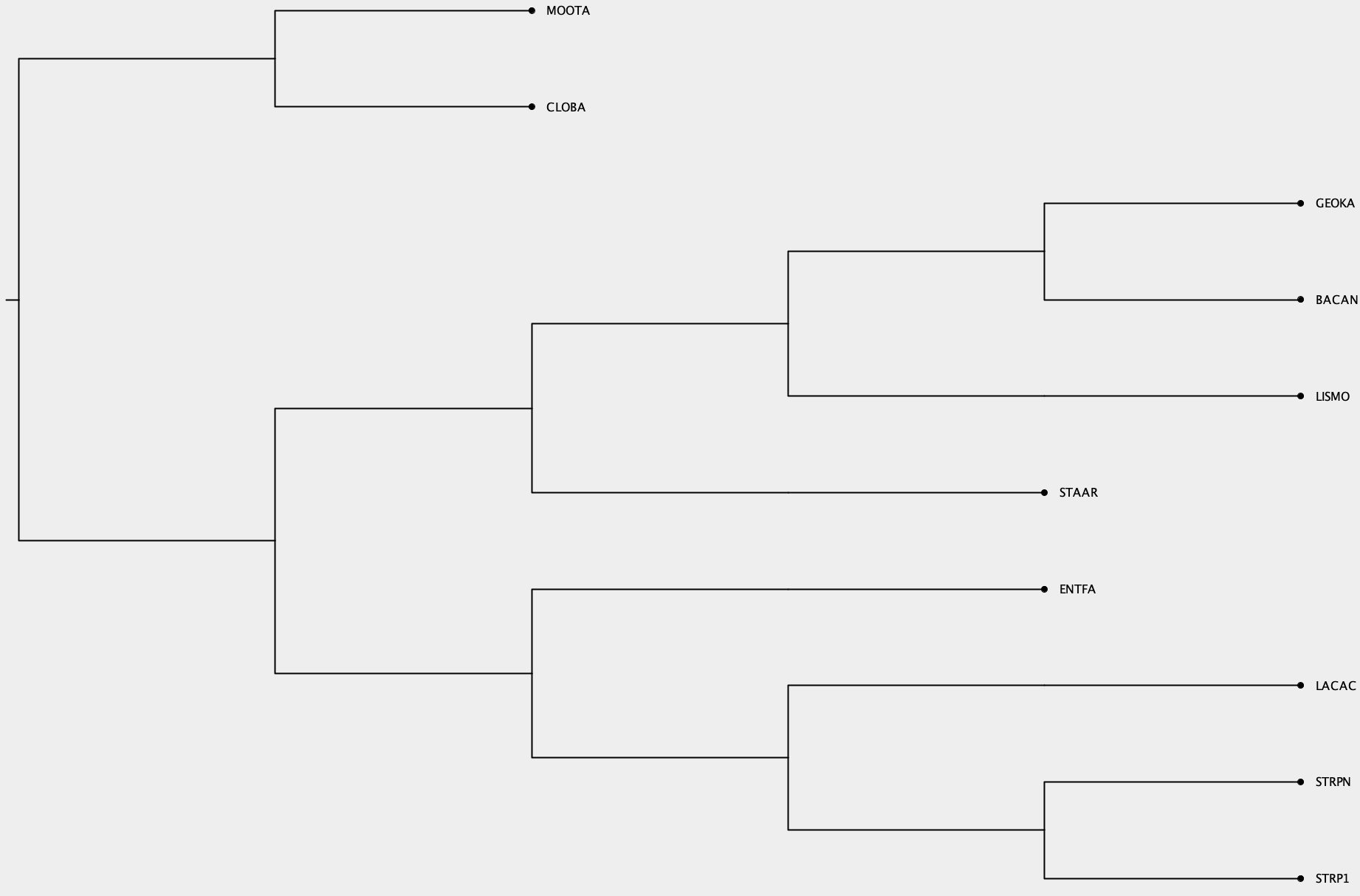
Нетривиальные ветви
NB! Мы называем ветвь нетривиальной, если она разбивает множество листьев на подмножества, в каждом из которых более одного элемента.
Я взяла 10 видов бактерий, дерево получилось большое, много нетривиальных ветвей (по формуле n-3=7).
| # | суть | какой таксон отделяет |
|---|---|---|
| 1 | {STRP1, STRPN} vs {LACAC, ENTFA, STAAR, LISMO, GEOKA, BACAN, MOOTA, CLOBA} | семейство Streptococcaceae |
| 2 | {STRP1, STRPN, LACAC} vs {ENTFA, STAAR, LISMO, GEOKA, BACAN, MOOTA, CLOBA} | |
| 3 | {STRP1, STRPN, LACAC, ENTFA} vs {STAAR, LISMO, GEOKA, BACAN, MOOTA, CLOBA} | порядок Lactobacillales |
| 4 | {STRP1, STRPN, LACAC, ENTFA, CLOBA, MOOTA} vs {STAAR, LISMO, BACAN, GEOKA} | порядок Bacillales |
| 5 | {STRP1, STRPN, LACAC, ENTFA, CLOBA, MOOTA, STAAR} vs {GEOKA, BACAN, LISMO} | |
| 6 | {STRP1, STRPN, LACAC, ENTFA, CLOBA, MOOTA, STAAR, LISMO} vs {BACAN, GEOKA} | семейство Bacillaceae |
| 7 | {STRP1, STRPN, LACAC, ENTFA, STAAR, LISMO, GEOKA, BACAN} vs {MOOTA, CLOBA} | класс Clostridia |
Задание 2 к практикуму 2. Сложность выбора
после долгих раздумий, функция белков: 16S рРНК-метилтрансфераза H (RSMH)
| # | название | мнемоника | SwissProt ID |
|---|---|---|---|
| 1 | Bacillus anthracis | BACAN | RSMH_BACAN |
| 2 | Clostridium botulinum | CLOBA | RSMH_CLOBA |
| 3 | Enterococcus faecalis | ENTFA | RSMH_ENTFA |
| 4 | Geobacillus kaustophilus | GEOKA | RSMH_GEOKA |
| 5 | Lactobacillus acidophilus | LACAC | RSMH_LACAC |
| 6 | Listeria monocytogenes serovar 1/2a | LISMO | RSMH_LISMO |
| 7 | Moorella thermoacetica | MOOTA | RSMH_MOOTA |
| 8 | Staphylococcus aureus | STAAR | RSMH_STAAR |
| 9 | Streptococcus pyogenes serotype M1 | STRP1 | RSMH_STRP1 |
| 10 | Streptococcus pneumoniae serotype 4 | STRPN | RSMH_STRPN |
Задание 3 к практикуму 2. Множественное выравнивание
задание
"Получите из Swiss-Prot последовательности белков с данной функцией из отобранных вами бактерий и выровняйте их."
куча способов но пожалуй первый
суть метода
1. Запуск JalView
2. File → Fetch sequences → "Select Database" → Uniprot
3. Через ; записать идентификаторы белков "xxxx_yyyyy" (xxxx – выбранная мнемоника функции, а yyyyy – мнемоники отобранных организмов)
4. OK
5. Web Service → Alignment → любая программа (например Muscle)

Задание 4 к практикуму 2
задание
"Отредактируйте названия последовательностей: оставьте от названия каждого белка только мнемонику вида (так легче будет сравнивать деревья). Если действовали через JalView, предварительно сохраните выравнивание в fasta-формате."
Задание 5 к практикуму 2. Реконструкция филогении
задание
"Откройте выравнивание в MEGA (методом "Analyze") и реконструируйте филогению тремя различными методами. Сохраните полученные деревья в формате Newick в файлы с такими названиями, по которым вы сможете потом понять, где какая реконструкция. (Замечание: чтобы MEGA открыла файл как выравнивание, он должен иметь расширение fasta или fa)."
UPGMA



Задание 7 к практикуму 2. fastme
задание
"Разберитесь, как пользоваться программой fastme на kodomo. Получите дерево и сравните результаты с результатом работы алгоритма Minimum evolution в программе MEGA. Опишите (в отчёте на отдельной странице), процедуру работы с программой и одинаковые ли получаются деревья (если нет, то попробуйте разобраться, почему)."
Программа FastME на кодомо позволяет быстро и точно построить филогению. Программа базируется на расстояниях (distance-based).
Синтаксис программы:
fastme [-i input data file] [-u input user tree file]
FastME можно также использовать и без аргументов.
Некоторые опции программы:
-i input data file- пользовательский файл с данными - выравнивание либо матрица расстояний
-u input user tree file- входной файл с филогенетическим деревом
-o output tree file- выходной файл с деревом, который запишет fastme
-O output matrix file- файл с матрицей расстояний, рассчитанных программой исходя из входного файла с выравниванием
-I output information file- выходной файл с данными о том, как сработала fastme
-B output bootstrap trees file- выходной файл с бутстреп-деревьями <><> <><>