В /nfs/srv/databases/ngs/esurikova/ были скопированы chr16.fasta и chr16.fastq.
Контроль качества чтений с помощью FastQC.
$ fastqc chr16.fastq Started analysis of chr16.fastq Approx 25% complete for chr16.fastq Approx 50% complete for chr16.fastq Approx 75% complete for chr16.fastq Analysis complete for chr16.fastq
Очистка чтений с помощью программы Trimmomatic. Параметры: порог качества = 20, длина чтений - не меньше 50.
$ java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr16.fastq chr16_trimmomatic.fastq TRAILING:20 MINLEN:50 TrimmomaticSE: Started with arguments: -phred33 chr16.fastq chr16_trimmomatic.fastq TRAILING:20 MINLEN:50 Automatically using 8 threads Input Reads: 3965 Surviving: 3798 (95,79%) Dropped: 167 (4,21%) TrimmomaticSE: Completed successfully
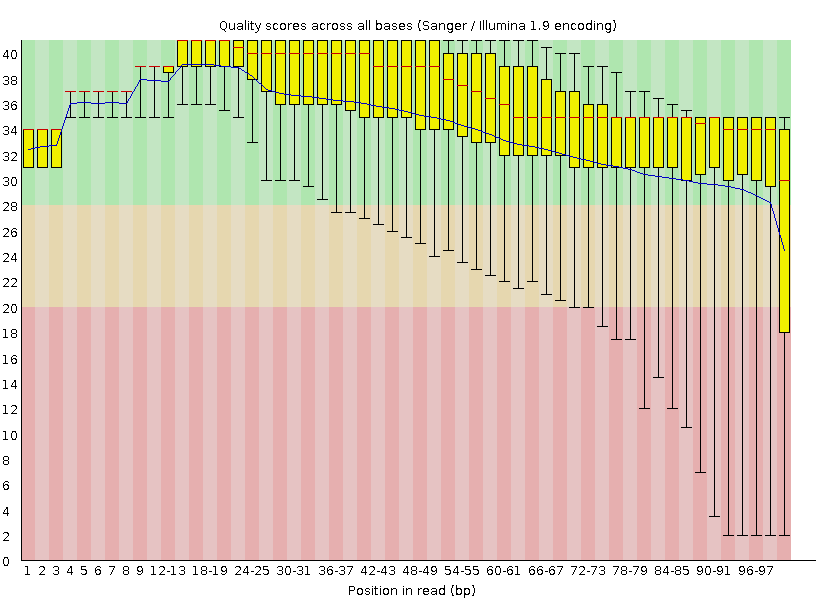
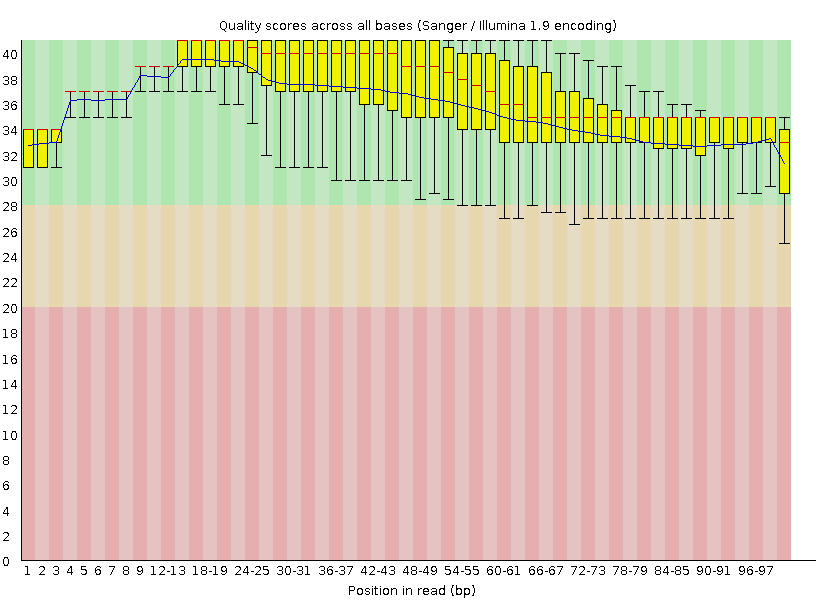
Был проведён анализ чтений FastQC после Trimmomatic. Сравнение показателей предсатвлено ниже.
| | |
| До - 3965 чтений. | После - 3798 чтений (95,79% от исходного кол-ва). |
Синяя линяя, отображающая среднее качество прочтений, на графике, оценивающем качество определeния нуклеотидов последовательности до использования Trimmomatic, значительно опускается по мере увеличения позиции нуклеотида в риде, нижние усики около 26% позиций опускаются в красную зону. После использования программы среднее качество снижается с ростом позиции менее существенно, интерквартильный размах целиком в зелёной зоне.
Индексирование референсной последовательности.
$ export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5
$ hisat2-build chr16.fasta chr16_indexed chr16_indexed.*.ht2 (* = 1, ..., 8)
Построение выравнивания прочтений и референса. Памаметры --no-spliced-alignment (выравнивние без разрывов) и --no-softclip (запрет подрезания ридов).
$ hisat2 -x chr16_indexed -U chr16_trimmomatic.fastq --no-spliced-alignment --no-softclip -S chr16_alignment.sam
3798 reads; of these:
3798 (100.00%) were unpaired; of these:
33 (0.87%) aligned 0 times
3763 (99.08%) aligned exactly 1 time
2 (0.05%) aligned >1 times
99.13% overall alignment rate
33 (0.87% от общего числа) рида не выровнены, 2 выровнены больше одного раза и 7763 (99.08%) выровнены один раз.
Перевод выравнивания в бинарный формат.
$ samtools view chr16_alignment.sam -b -o chr16_alignment.bam
Сортировка выравнивания по координате в референсе начала чтения.
$ samtools sort chr16_alignment.bam -T chr16_alignment_temp.txt -o chr16_alignment_sort.bam
Индексирование полученного файла.
$ samtools index chr16_alignment_sort.bam
Анализ: 3770 ридов картировано, некартированных нет.
$ samtools idxstats chr16_alignment_sort.bam chr16 90354753 3770 0 * 0 0 33
Создание файла с полиморфизмами.
$ samtools mpileup -uf chr16.fasta chr16_alignment_sort.bam -o chr16_snp.bcf [fai_load] build FASTA index. [mpileup] 1 samples in 1 input filesSet max per-file depth to 8000
Создание файла со списком отличий между референсом и чтениями в формате .vcf.
$ bcftools call -cv chr16_snp.bcf -o chr16_snp.vcf There are total 65 snps, 64 replacements, 1 INDEL.
Ниже таблица с примерами полиморфизмов.
| CHROM | TYPE OF SNP | ID | REF | READ | QUAL | DEPTH |
| chr16 | Indel | 11444454 | gaaaaaaaaaaa | gaaaaaaaaaa,gaaaaaaaaa | 10.8299 | 55 |
| chr16 | Replasing | 11444469 | C | T | 221.999 | 92 |
| chr16 | Replasing | 11444572 | A | C | 225.009 | 113 |
Конвертация файла (без инделей) в .avinput и аннотирование по базам данных: refgene, dbsnp, 1000 genomes, GWAS, Clinvar.
$ perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 chr16_snp_without_indel.vcf -o chr16.avinput NOTICE: Finished reading 96 lines from VCF file NOTICE: A total of 64 locus in VCF file passed QC threshold, representing 64 SNPs (38 transitions and 26 transversions) and 0 indels/substitutions NOTICE: Finished writing 64 SNP genotypes (38 transitions and 26 transversions) and 0 indels/substitutions for 1 sample $ perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr16_snp_rs -build hg19 -dbtype snp138 chr16.avinput /nfs/srv/databases/annovar/humandb/ NOTICE: Variants matching filtering criteria are written to chr16_snp_rs.hg19_snp138_dropped, other variants are written to chr16_snp_rs.hg19_snp138_filtered NOTICE: Processing next batch with 64 unique variants in 64 input lines NOTICE: Database index loaded. Total number of bins is 2894320 and the number of bins to be scanned is 38 NOTICE: Scanning filter database /nfs/srv/databases/annovar/humandb/hg19_snp138.txt...Done chr16_snp_rs.hg19_snp138_dropped chr16_snp_rs.hg19_snp138_filtered chr16_snp_rs.log $ perl /nfs/srv/databases/annovar/annotate_variation.pl -out chr16_refgene -build hg19 chr16.avinput /nfs/srv/dat abases/annovar/humandb/ NOTICE: The --geneanno operation is set to ON by default NOTICE: Reading gene annotation from /nfs/srv/databases/annovar/humandb/hg19_refGene.txt ... Done with 50914 transcripts (including 11516 without coding sequence annotation) for 26271 unique genes NOTICE: Reading FASTA sequences from /nfs/srv/databases/annovar/humandb/hg19_refGeneMrna.fa ... Done with 9 sequences WARNING: A total of 345 sequences will be ignored due to lack of correct ORF annotation NOTICE: Finished gene-based annotation on 64 genetic variants in chr16.avinput NOTICE: Output files were written to chr16_refgene.variant_function, chr16_refgene.exonic_variant_function chr16_refgene.exonic_variant_function chr16_refgene.log chr16_refgene.variant_function $ perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype snp138 -out chr16_dbsnp -build hg19 chr16.avinput /nfs/srv/databases/annovar/humandb/ NOTICE: Variants matching filtering criteria are written to chr16_dbsnp.hg19_snp138_dropped, other variants are written to chr16_dbsnp.hg19_snp138_filtered NOTICE: Processing next batch with 64 unique variants in 64 input lines NOTICE: Database index loaded. Total number of bins is 2894320 and the number of bins to be scanned is 38 NOTICE: Scanning filter database /nfs/srv/databases/annovar/humandb/hg19_snp138.txt...Done chr16_dbsnp.hg19_snp138_dropped chr16_dbsnp.hg19_snp138_filtered chr16_dbsnp.log $ perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype 1000g2014oct_all -out chr_1000genomes -buildver hg19 chr16.avinput /nfs/srv/databases/annovar/humandb/ NOTICE: Variants matching filtering criteria are written to chr_1000genomes.hg19_ALL.sites.2014_10_dropped, other variants are written to chr_1000genomes.hg19_ALL.sites.2014_10_filtered NOTICE: Processing next batch with 64 unique variants in 64 input lines NOTICE: Database index loaded. Total number of bins is 2824642 and the number of bins to be scanned is 38 NOTICE: Scanning filter database /nfs/srv/databases/annovar/humandb/hg19_ALL.sites.2014_10.txt...Done chr_1000genomes.hg19_ALL.sites.2014_10_dropped chr_1000genomes.hg19_ALL.sites.2014_10_filtered chr_1000genomes.log $ perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -dbtype gwasCatalog -out chr16_gwas -buildver hg19 chr16.avinput /nfs/srv/databases/annovar/humandb/ NOTICE: Reading annotation database /nfs/srv/databases/annovar/humandb/hg19_gwasCatalog.txt ... Done with 18027 regions NOTICE: Finished region-based annotation on 64 genetic variants in chr16.avinput NOTICE: Output file is written to chr16_gwas.hg19_gwasCatalog chr16_gwas.hg19_gwasCatalog chr16_gwas.log $ perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype clinvar_20150629 -out chr16_clinvar -buildver hg19 chr16.avinput /nfs/srv/databases/annovar/humandb/ NOTICE: the --dbtype clinvar_20150629 is assumed to be in generic ANNOVAR database format NOTICE: Variants matching filtering criteria are written to chr16_clinvar.hg19_clinvar_20150629_dropped, other variants are written to chr16_clinvar.hg19_clinvar_20150629_filtered NOTICE: Processing next batch with 64 unique variants in 64 input lines NOTICE: Database index loaded. Total number of bins is 49139 and the number of bins to be scanned is 1 NOTICE: Scanning filter database /nfs/srv/databases/annovar/humandb/hg19_clinvar_20150629.txt...Done chr16_clinvar.hg19_clinvar_20150629_dropped chr16_clinvar.hg19_clinvar_20150629_filtered chr16_clinvar.log
Базы данных:
За ислючением clinvar'а базы данных выдали аннотации. _dropped - аннотированное, _filtered - неаннотированное.
Расположение SNP
| Расположение | Число SNP | Пояснение |
| интронное | 23 | вариант в интроне |
| экзонное | 9 | вариант в экзоне |
| 3'-НТО | 3 | вариант в 3'-нетранслируемой области |
| 5'-НТО | 3 | вариант в 5'-нетранслируемой области |
| интергенное | 19 | вариант между генами |
| downstream | 2 | вариант в 1-kb области ниже сайта окончания транскрипции |
SNP в экзонах
| Координаты | Ген | экзон:замена нуклеотид:замена | Референс | Рид | Тип замены | Качество | Глубина рида | Генотип |
| 11362729 | TNP2:NM_005425 | exon1:c.C391T:p.R131W | G | A | несинонимичная | 225.009 | 145 | гетеро |
| 11367143 | PRM3:NM_021247 | exon1:c.C310T:p.R104X | G | A | нонсенс | 221.999 | 35 | гомо |
| 11367154 | PRM3:NM_021247 | exon1:c.G299A:p.R100Q | C | T | несинонимичная | 221.999 | 30 | гомо |
| 11369938 | PRM2:NM_001286359 | exon1:c.C290T:p.P97L | G | A | несинонимичная | 125.008 | 27 | гетеро |
| 11369947 | PRM2:NM_001286359 | exon1:c.C281T:p.A94V | G | A | несинонимичная | 139.008 | 27 | гетеро |
| 11374866 | PRM1:NM_002761 | exon2:c.C139A:p.R47R | G | T | несинонимичная | 221.999 | 38 | гомо |
| 11444572 | RMI2:NM_152308 | exon2:c.A369C:p.T123T | A | C | несинонимичная | 225.009 | 113 | гетеро |
| 31096164 | PRSS53:NM_001039503 | exon8:c.C1216G:p.P406A | G | G | несинонимичная | 148.077 | 7 | гетеро |
| 56969148 | HERPUD1:NM_014685 | exon2:c.G149A:p.R50H | G | A | несинонимичная | 225.009 | 80 | гетеро |
Заболевания, вызванные SNP
| Заболевание | Хромосома | Координата | Референс | Рид | Генотип | Качество | Глубина рида |
| Черты, связанные с ожирением | chr16 | 11374866 | G | T | гомо | 221.999 | 38 |
| Болезнб Паркинсона | chr16 | 31095171 | C | T | гомо | 221.999 | 53 |
| Метаболический синдром | chr16 | 56969148 | G | A | гетеро | 225.009 | 80 |
Таблица со всей информацией, полученной из аннотаций
| Команда | Значение | fastqc chr16.fastq | запуск fastaqc - контроль качества чтений | java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr16.fastq chr16_trimmomatic.fastq TRAILING:20 MINLEN:50 | очистка чтений, порог качества - 20, минимальная длина - 50 | export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5 | экспорт hisat2 | hisat2-build chr16.fasta chr16_indexed chr16_indexed.*.ht2 (* = 1, ..., 8) | индексирование референсной последовательности | hisat2 -x chr16_indexed -U chr16_trimmomatic.fastq --no-spliced-alignment --no-softclip -S chr16_alignment.sam | построение выравниваний между прочтениями и референсом без обрезки концов без разрывов | samtools view chr16_alignment.sam -b -o chr16_alignment.bam | перевод выравнивания в бинарный формат | samtools sort chr16_alignment.bam -T chr16_alignment_temp.txt -o chr16_alignment_sort.bam | сортировка выравнивания прочтений с референсом по координате референса начала чтения | samtools index chr16_alignment_sort.bam | индексирование отсортированного выравнивания | samtools idxstats chr16_alignment_sort.bam | анализ картированности прочтений | samtools mpileup -uf chr16.fasta chr16_alignment_sort.bam -o chr16_snp.bcf | создание файла с полиморфизмами | bcftools call -cv chr16_snp.bcf -o chr16_snp.vcf | список отличий между референсом и прочтениями | perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 chr16_snp_without_indel.vcf -o chr16.avinput | конвертация файла | perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr16_snp_rs -build hg19 -dbtype snp138 chr16.avinput /nfs/srv/databases/annovar/humandb/ | аннотация по бд snp | perl /nfs/srv/databases/annovar/annotate_variation.pl -out chr16_refgene -build hg19 chr16.avinput /nfs/srv/dat | аннотация по бд refgene | perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype 1000g2014oct_all -out chr_1000genomes -buildver hg19 chr16.avinput /nfs/srv/databases/annovar/humandb/ | аннотация по бд 1000genomes | perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -dbtype gwasCatalog -out chr16_gwas -buildver hg19 chr16.avinput /nfs/srv/databases/annovar/humandb/ | аннотация по бд gwas | perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype clinvar_20150629 -out chr16_clinvar -buildver hg19 chr16.avinput /nfs/srv/databases/annovar/humandb/ | аннотация по бд clinvar |