На главную страницу четвертого семестра
They drew long and elaborate family-trees with unnumerable branches.
J.R.R.Tolkien "The Lord of the Rings", Prologue, "Concerning Hobbits".
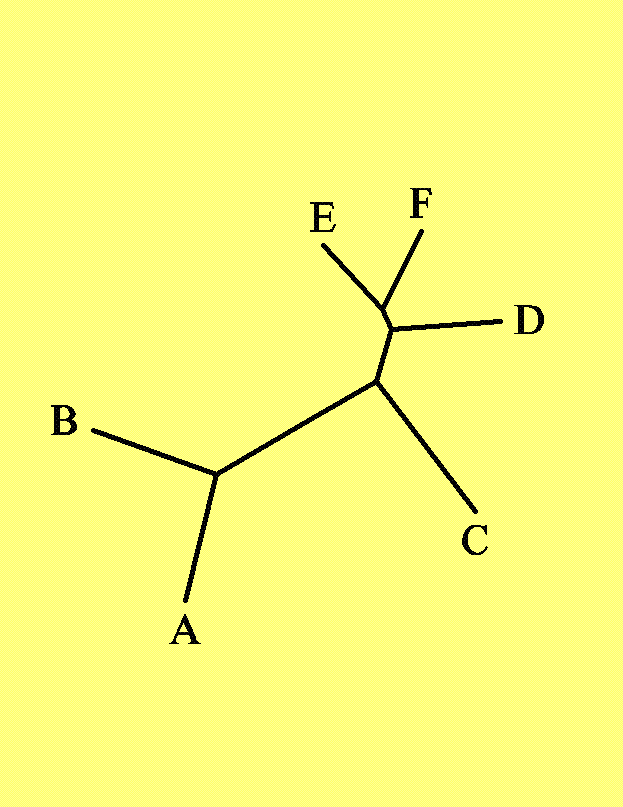
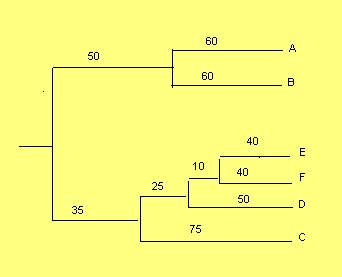
Исходным материалом для наших упражнений будет дерево, заданное следующей скобочной формулой. Ниже приведено его графическое изображение.((А:60,В:60):50,(((Е:40,F:40):10,D:50):25,С:75):35);
Из того, что суммы по длинам ветвей для каждого листа одинаковы, мы делаем вывод, что сравнительная степень расхождения одинакова (т.е. если мы принимаем гипотезу молекулярных часов, можно считать, что одинаково время, прошедшее с момента расхождения наших последовательностей). Следует заметить, что т.к. дерево ультраметрично, то оно по умолчанию является укорененным. Далее мы воспользуемся этим.
| A | B | C | D | E | F | |
| 1 | * | * | . | . | . | . |
| 2 | * | * | * | * | . | . |
| 3 | * | * | * | . | . | . |
N=4*n(AB)*L/3*100
Т.е. выраженную в процентах сходства длину ветки AB (n(AB)/100) умножаем на длину гена L и на коэффициент 4/3 (он введен, чтобы получить именно то число замен, которое нам нужно, иначе из-за несовершенства алгоритма msbar, который в каждом четвертом случае меняет нуклеотид на самого себя, считая это заменой, реальное число будет составлять только 75% от нужного). См.предыдущее задание. Мутантные последовательности получены с помощью следующего скрипта.msbar frc.fasta ab.fasta -count 835 -point 4 -auto msbar frc.fasta efdc.fasta -count 585 -point 4 -auto msbar ab.fasta a.fasta -count 1000 -point 4 -auto msbar ab.fasta b.fasta -count 1000 -point 4 -auto msbar efdc.fasta efd.fasta -count 415 -point 4 -auto msbar efdc.fasta c.fasta -count 1250 -point 4 -auto msbar efd.fasta ef.fasta -count 165 -point 4 -auto msbar efd.fasta d.fasta -count 830 -point 4 -auto msbar ef.fasta e.fasta -count 665 -point 4 -auto msbar ef.fasta f.fasta -count 665 -point 4 -auto cat a.fasta > mutants.fasta cat b.fasta >>mutants.fasta cat c.fasta >>mutants.fasta cat d.fasta >>mutants.fasta cat e.fasta >>mutants.fasta cat f.fasta >>mutants.fasta |
IДерево, полученное алгоритмом Neighbor-joining, мспользующим матрицу попарных расстояний. Не совсем такое дерево, как было. Алгоритм не предусматривает молекулярные часы и строит неукорененное дерево. Сущность алгоритма в последовательном объединении объектов по принципу наибольшего сходства. Топология обсуждается ниже.
+--------B ! ! +---------------------C ! +-2 ! ! +----------D 1--------------------3 ! ! +------E ! +-4 ! +-------F ! +--------------AII Дерево, полученное алгоритмом UPGMA, использующим матрицу попарных расстояний. В отличие от предыдущего, укорененное и ультраметрическое (алгоритм предусматривает молекулярные часы). Оно не отличается по топологии от истинного дерева
+------a
+-----3
! +------b
--5
! +--------c
+---4
! +---d
+----2
! +-e
+-1
+-f
III Дерево, полученное алгоритмом максимального правдоподобия (программа fdnaml, параметр -ttratio 1). Что мы знаем про этот алгоритм? Это метод символьно-ориентированный, использующий не матрицу расстояний, а модель эволюции. Он может как содержать, так и не содержать предположение молекулярных часов. В данном случае не содержит, поэтому дерево неультраметрическое и неукорененное. На истинное оно не похоже.
+---------D
|
+--------------------------1 +---E
| | +---2
| +--3 +-------------------------C
| |
| +-----F
|
4-----B
|
+---------------A
Табл.2 Сравнение деревьев, построенных разными алгоритмами.
|
|
A |
B |
C |
D |
E |
F |
Исходное |
NJ |
UPGMA |
Макс. пр. |
|
1 |
* |
* |
. |
. |
. |
. |
+ |
+ |
+ |
+ |
|
2 |
* |
* |
* |
* |
. |
. |
+ |
- |
+ |
- |
|
3 |
* |
* |
* |
. |
. |
. |
+ |
+ |
+ |
- |
|
4 |
. |
. |
* |
* |
. |
. |
- |
+ |
- |
- |
|
5 |
* |
* |
. |
* |
. |
. |
- |
- |
- |
+ |
|
6 |
* |
* |
. |
* |
. |
* |
- |
- |
- |
+ |
+---------------------------D
|
| +------A
| +-------100.0-|
| | +------B
+------|
| +------E
| +-33.0-|
+-30.0-| +------C
|
+-------------F
Дерево не похоже на реальное, единственная внутренняя ветвь, встречающаяся и в том, и в другом, имеет бутстреп-значение 100, т.е. является абсолютно достоверной.
Это ветвь [AB]. Появились 2 внутренние ветви, достоверность которых крайне сомнительна - их значения 33 и 30. Это ветви [CE] и [ABD]. В то же время ветви истинного дерева, не попавшие в бутстреп-реконструкцию, имеют значения 28 и 18 (не намного меньше).
Ветвь [EF] получила значение 28, а [ABC] - только 18. Можно предположить, что такие результаты связаны с недостаточностью числа реплик - 100 недостаточно для статистической достоверности.
А как же выглядело исходное дерево? Исходная скобочная формула была визуализирована с помощью программы drawtree. Полученный рисунок неукорененного дерева можно видеть ниже.Это филограмма, дерево, сохраняющее расстояние между объектами.