Анализ результатов поиска по профилю
Разделение выравнивания представителей домена, построенного в практикуме 9, на две группы
Разделяю полученное ранее выравнивание по доменной архитектуре белков. Такое разделение позволяет получить 2 четко отличающиеся группы, которым сооветствуют наилучшие выравнивания.
Анализируя таксономию организмов, я рассматривала 7 таксонов с разным числом представителей. Каждый таксон содержит как представителей первой архитектуры, так и представителей второй. Консервативность и сходство последовательностей внутри архитектуры выше. При выборе двух таксонов, последовательности относились бы как к первой, так и ко второй архитектуре, а, следовательно, были бы менее консервативны и схожи между собой. что .
Деление последовательностей на основании анализа дерева также ухудшило бы выравнивание, т.к. среди последовательностей со второй архитектурой оказалась бы одна последовательность с первой.
Построение профиля, отличающего одну группу последовательностей от другой
Исходные данные: два выравнивания последовательностей домена - arch1.fasta - соответствует первой группе, и arch2.fasta - сооветствует второй группе.
Подготавливаю выравнивание в формате MSF.
Добавляю веса последовательностей в выравнивания:
Создаю профили arch1.prf, arch2.prf:
Подготавливаю файл с последовательностями из обеих групп, по которому осуществляется поиск.
Этап нормализации для данной задачи опускается.
Провожу поиск по профилям среди последовательностей обеих групп. Порог веса ставлю маленьким (-C 0.0), чтобы все последовательности оказались в выдаче.
Строю графики весов находок pfsearch, отсортированных по убыванию, для первой и второй группы соответственно:
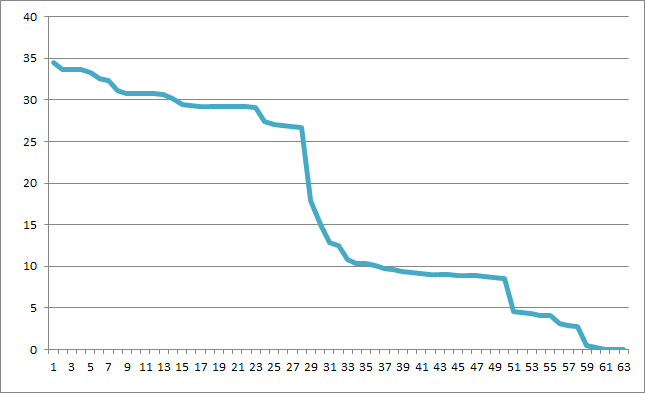
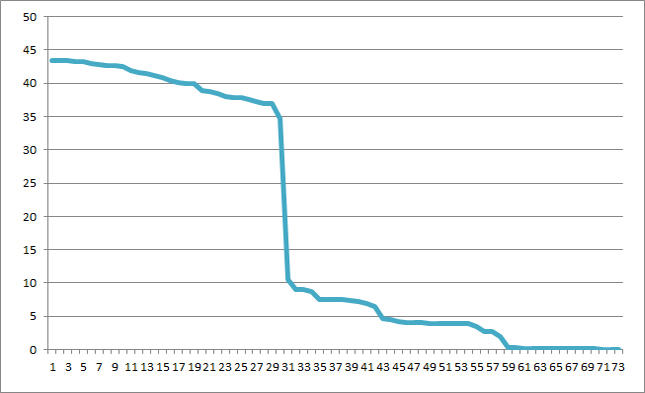
Ступеньку на графиках можно интерпретировать как порог нормализованного веса для находок из группы.
Строю ROC-кривые. Правильные находки - те, которые принадлежат группе, по которой построен профиль. За исключение принимаю последовательности, которым соответствует более одной находки (2 или 3 - по разным участкам последовательности), последние из которых имеют малое значение нормализованного веса (не более 3.10).
Ниже изображены кривые для первой и второй группы соответственно:
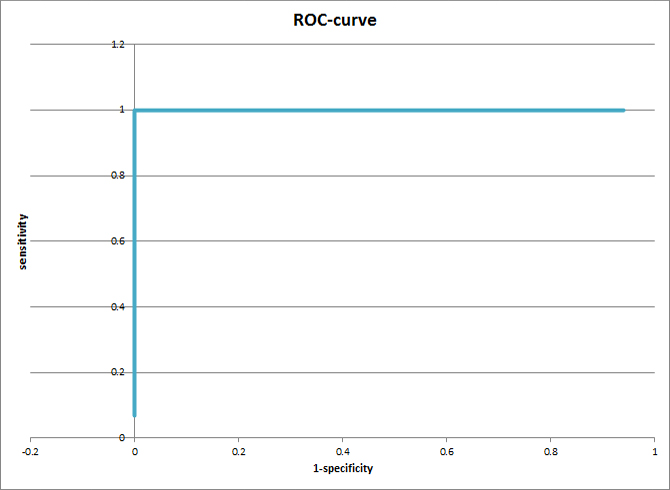
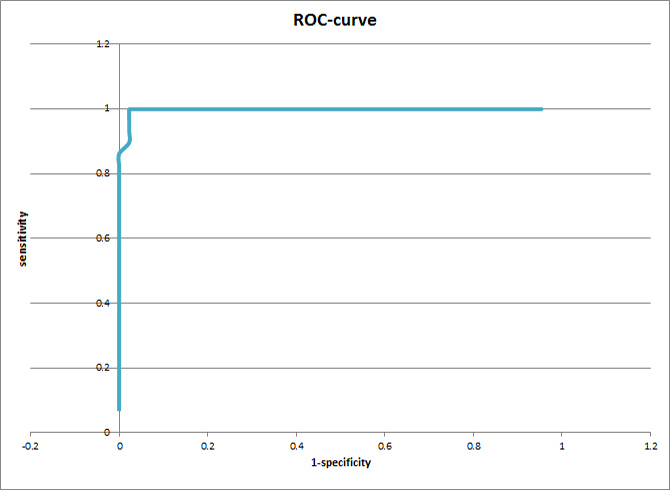
На основе анализа ROC-кривых для первой группа можно предложить порог 17.87 - дает 0 ошибок первого и второго рода, для второй - 34.65, дающий 0 ошибок первого рода и 1 ошибку второго рода (с вероятностью 0.033 - 1 ошибка из 30).
Привожу таблицы с описанием находок для первой и второй группы соответственно:
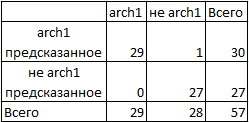
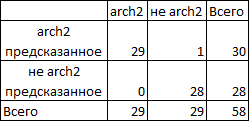
Для первой группы итоговое число последовательностей получилось на 1 меньше исходного. Я полагаю, что это связанно с очень малым значением нормализованного веса для последовательности A3QCN7_SHELP. Действительно, если посмотреть на филогенетическое дерево, полученное в практикуме 10, то видно, что данный белок наиболее близок к белку A3D6P5_SHEB5 с минимальным из приведенных нормализованных весов - 0.01.
Таким образом, полученные профили позволяют отличить заданные группы последовательностей без ошибок для первой группы и с допустимой погрешнотью для второй.
Все полученные результаты приведены в arch1.xls и arch2.xls.
© Eugenia Prokhorova 2011