Филогенетические деревья. Занятие 4. Реконструкция деревьев по нуклеотидным последовательностям. Анализ деревьев, содержащих паралоги
1. Построение дерева по нуклеотидным последовательностям
Строю филогенетическое дерево тех же бактерий, что в предыдущих заданиях, используя последовательности РНК малой субъединицы рибосомы (16S rRNA). Нужные последовательности вырезаю из записей EMBL с помощью seqret с опцией -sask.
| Название | Мнемоника | AC записи EMBL | Координаты 16S рРНК | Цепь |
| Bacillus subtilis | BACSU | AL009126 | 9810-11364 | прямая |
| Clostridium tetani | CLOTE | AE015927 | 8715-10223 | прямая |
| Geobacillus kaustophilus | GEOKA | BA000043 | 10421-11973 | прямая |
| Lactobacillus acidophilus | LACAC | CP000033 | 59255-60826 | прямая |
| Lactococcus lactis | LACLM | CP002365 | 470580-472133 | прямая |
| Listeria monocytogenes | LISMO | AL591974 | 37466-39020 | прямая |
| Staphylococcus aureus | STAA1 | CP003808 | 540419-542060 | прямая |
Осуществляю выравнивание Muscle with Defaults в JalView, используя Web Service. Получаю allalign.fasta. Провожу бутстрэп-анализ, используя метод, доступный в программе MEGA - Neighbour Joining. В окошке, которое открывается после вызова программы, в меню "Test of Phylogeny" выбираю "Bootstrap method". Указываю число реплик, равное 100.
Original tree и Bootstrap consensus tree соответственно
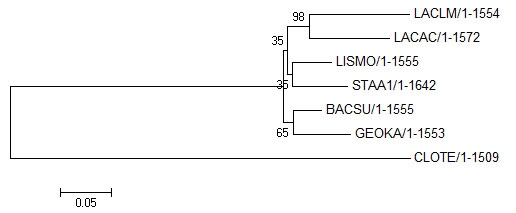
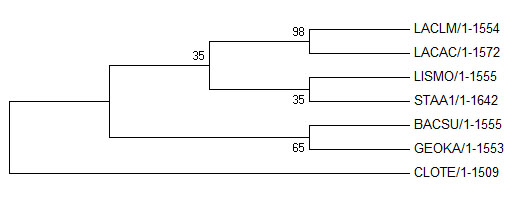
Правильное дерево:
Полученное дерево не совпадает с правильным.
Новые ветви: {LACLM, LACAC, LISMO, STAA1} против {BACSU, GEOKA, CLOTE}, {LISMO, STAA1} против {LACLM, LACAC, BACSU, GEOKA, CLOTE}. Эти ветви ошибочны и имеют наименьшую поддержку - по 35, что сразу говорит о большой вероятности ошибки при определении их положения в дереве.
Отсутствующие ветви: {BACSU, GEOKA, LISMO} против {CLOTE, LACAC, LACLM, STAA1}, {BACSU, GEOKA, LISMO, STAA1} против {CLOTE, LACAC, LACLM}.
Далее пробую получить выравнивание с Clustal. Получаю allalign2.fasta. Снова провожу бутстрэп-анализ с Neighbour Joining.
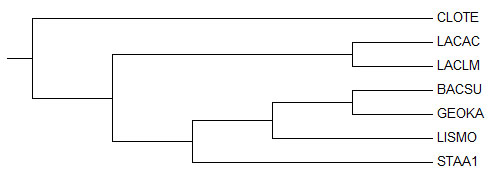
Original tree и Bootstrap consensus tree соответственно
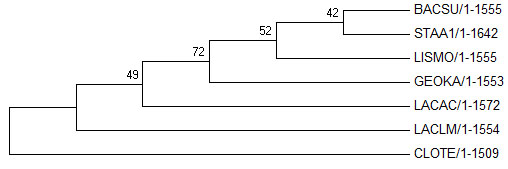
Правильное дерево:
Полученное дерево снова не совпадает с правильным.
Новые ветви: {BACSU, STAA1} против {LISMO, GEOKA, LACAC, LACLM, CLOTE}, {BACSU, STAA1, LISMO} против {GEOKA, LACAC, LACLM, CLOTE}, {BACSU, STAA1, LISMO, GEOKA, LACAC} против {LACLM, CLOTE}.
Отсутствующие ветви: {LACAC, LACLM} против {CLOTE, BACSU, GEOKA, LISMO, STAA1}, {BACSU, GEOKA} против {CLOTE, LACAC, LACLM, LISMO, STAA1}, {BACSU, GEOKA, LISMO} против {CLOTE, LACAC, LACLM, STAA1}.
Удаление некоторых "плохих" участков выравнивания приводит к аналогичным деревьям (с теми же нетривиальными ветвями). Качество реконструкции по сравнению с деревьями, построенными по белкам оказалось ниже (дерево, полученное данным методом на прошлом занятии совпало с правильным).
Мне казалось, белковые выравнивания предпочтительнее для исследований, так или иначе связанных с функциями молекул, т.к. они позволяют учесть свойства аминокислотных остатков. Очевидно, вероятность случайного совпадения при анализе нуклеотидных последовательностей меньше, т.к. стандартных нуклеотидов в 5 раз меньше, чем аминокислот, и многие аминокислоты кодируются несколькими кодонами (до 6). Т.е. нуклеотидные выравнивания позволяют увидеть более реальную картину гомологии. Плюс, исследуя филогенетику с помощью рРНК, наверное, неплохо бы учитывать вторичную структуру. Она содержит много шпилек и довольно сурова, можно посмотреть.
Впринципе, при построении филогенетических деревьев организмов, я бы использовала и белковые и нуклеотидные последовательности, т.к. для их выравниваний есть свои преимущества и свои недостатки.
2. Построение и анализ дерева, содержащего паралоги
Нахожу в своих бактериях достоверные гомологи белка CLPX_BACSU и строю дерево этих гомологов. Чтобы найти гомологов в заданных организмах, использую файл proteo.fasta с диска P, в котором лежат записи банка UNIPROT, относящиеся к бактериям, перечисленным в таблице к заданию 1. Провожу поиск программой blastp гомологов (с порогом на E-value = 0,001) и отбираю по мнемонике видов только те находки, которые относятся к отобранным бактериям - clpx.txt. Получаю hom.fasta.
Осуществляю выравнивание Muscle with Defaults в JalView. Получаю homal.fasta. Провожу бутстрэп-анализ, используя Neighbour Joining.
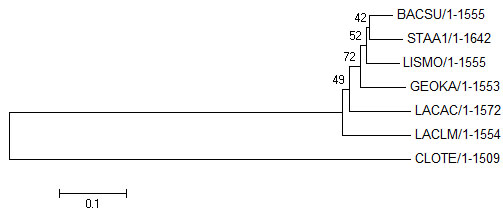
Bootstrap consensus tree

Два гомологичных белка - ортологи, если они а) из разных организмов; б) разделение их общего предка на линии, ведущие к ним, произошло в результате видообразования.
Примеры: CLPX_BACSU и CLPX_GEOKA, HSLU_STAA1 и HSLU_LISMO.
Два гомологичных белка из одного организма - паралоги.
Примеры: Q8Y8B1_LISMO и Q8YAB6_LISMO, Q891B9_CLOTE и Q899H3_CLOTE.
© Eugenia Prokhorova 2011