Cеми-эмпирические вычисления: Mopac
1. Структура порфирина
Была найдена аннотация порфирина в виде SMILES и создан текстовый файл 1.smi, где сначала указан SMILES порфирина, а через несколько пробелов просто porphyrin. На основе этого описания c помощью программы из openbabel вызовом из cmd была построена 3D структура порфирина: obgen 1.smi > 1.mol.
Полученная структура (рис. 1) была просмотрена в PyMol для удаления потенциально ненужных водородов, результат (рис. 2) был сохранен в формате pdb.
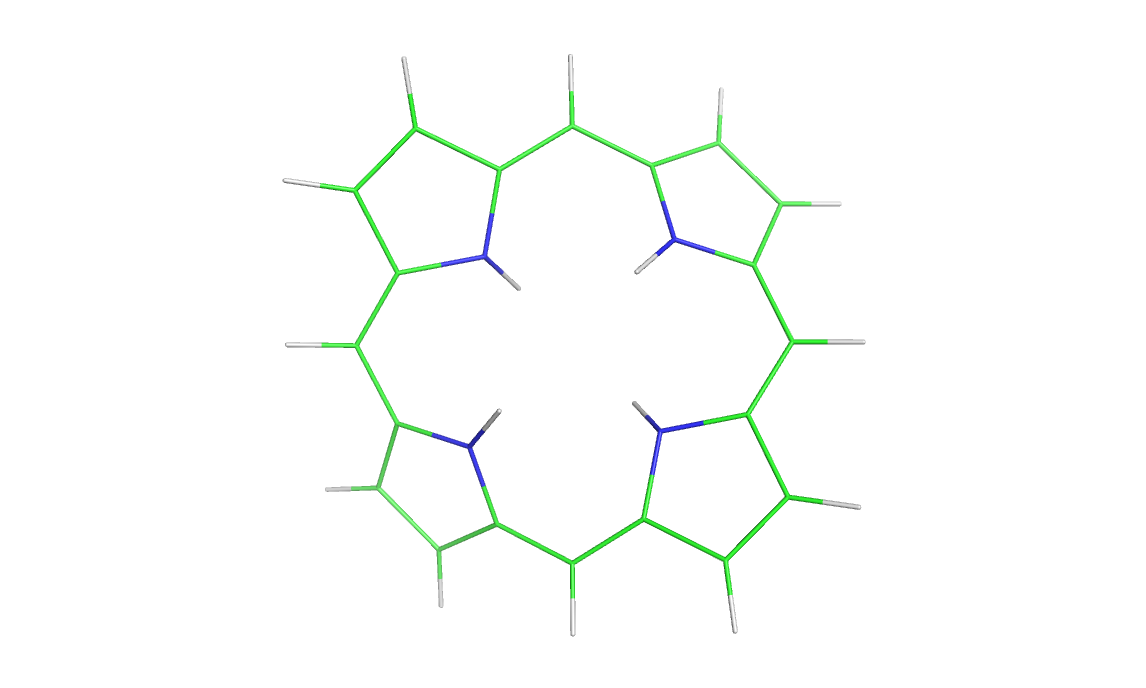
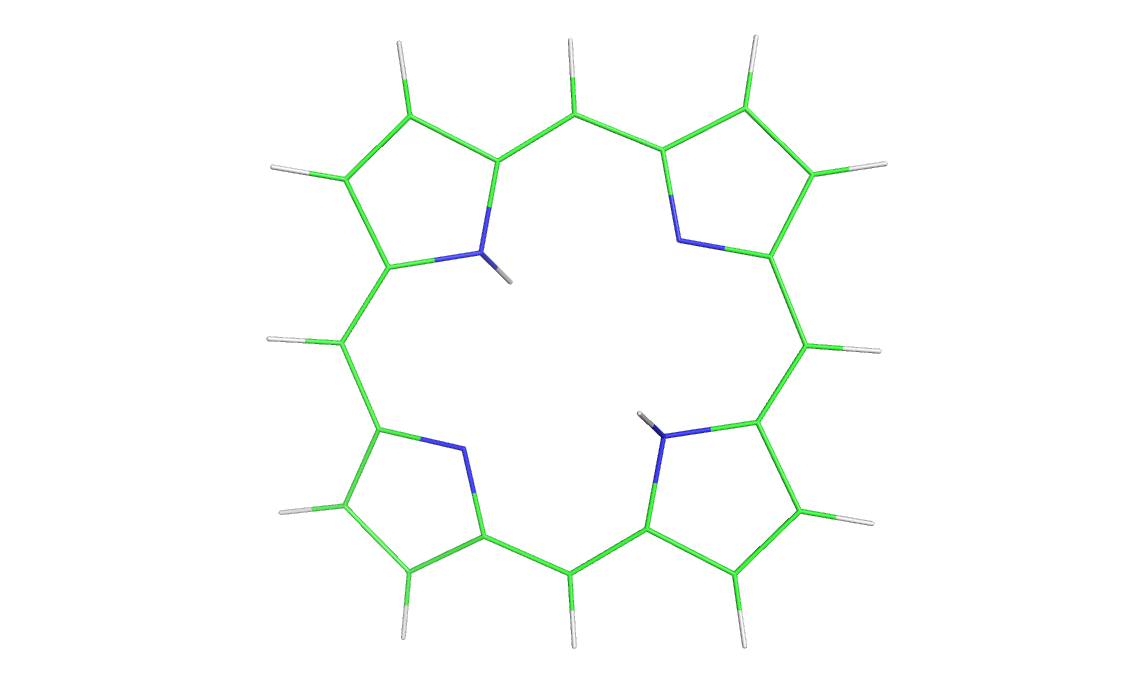
Рис. 1. Изначальный вариант структуры. Рис. 2. Структура после удаления лишних водородов.
С помощью Openbabel осуществлено переформатирование координат в mol формате во входной файл для Mopac: babel -ipdb porph_ed.pdb -omop 1_opt.mop -xk "PM6" (-xk задает тип параметризации pm6).
Запуск Mopac проводился на kodomo командой MOPAC2009.exe 1_opt.mop. Результат 1_opt.out был переформатирован в pdb: babel -imopout 1_opt.out -opdb 1_opt.pdb и визуализирован в PyMol. В отличие от исходной молекулы (рис. 3), молекула после Mopac имеет плоскую конформацию (рис. 4).
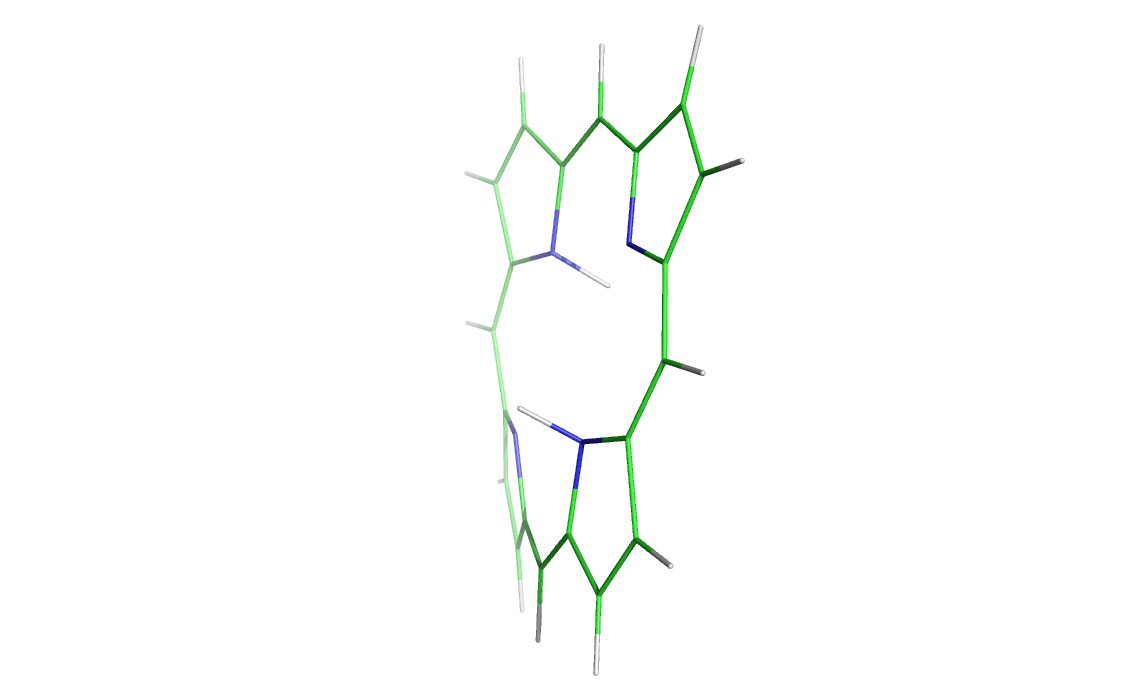
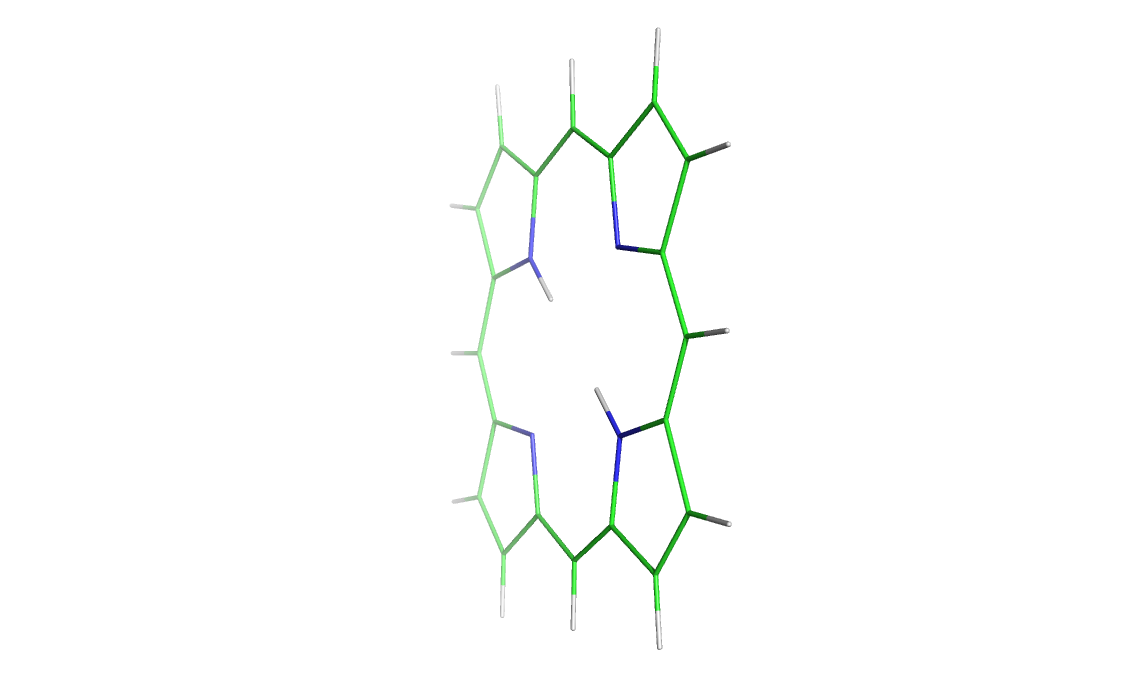
Рис. 3. Структура до оптимизации. Рис. 4. Структура порфирина после оптимизации.
2. Возбужденные состояния порфирина
Проведен рассчет возбужденных состояний порфирина и на основе этих данных был оценен спектр поглощения молекулы. Для расчёта возбуждённых состояний сделана копия mop файла из предыдущего задания - 1_opt_spectr.mop. Для указания Mopac о необходимости расчёта возбуждённого состояния в конец файла добавлялось: cis c.i.=4 meci oldgeo; Additional data. Далее был запущен Mopac.
В конце выходного файла были найдены значения энергий для электронных переходов:
STATE ENERGY (EV) Q.N. SPIN SYMMETRY POLARIZATION
ABSOLUTE RELATIVE X Y Z
1+ 0.000000 0.000000 1+ SINGLET ????
2 1.914650 1.914650 1 TRIPLET ????
3 2.267178 2.267178 2 SINGLET ????
4 2.464890 2.464890 2 TRIPLET ????
5 2.825739 2.825739 3 TRIPLET ????
6 3.364665 3.364665 4 TRIPLET ????
7 3.391936 3.391936 3 SINGLET ???? 0.2018 0.2373 0.0011
8 3.669748 3.669748 4 SINGLET ???? 2.3998 2.0267 0.0129
9 3.871564 3.871564 5 SINGLET ???? 1.5364 1.7970 0.0087
Перевод данных значении энергий переходов в джоулях осуществлялся умножением на 1,6x10-19. C использованием формулы E=hv=hc/λ был произведен рассчет длин волн, при которых происходят эти переходы.
Полученные значения говорят о поглощении порфирина в ближнем ультрафиолете и в видимой части спектра:
2 320 nm
3 337 nm
4 365 nm
5 368 nm
6 438 nm
7 503 nm
8 546 nm
9 647 nm
3. Геометрия молекул
Для молекулы бензохинона O=C1C=CC(=O)C=C1 выполением шагов, описанных в задании 1, были получены структуры, представленные на рис. 5 (obgen) и 6 (Mopac).
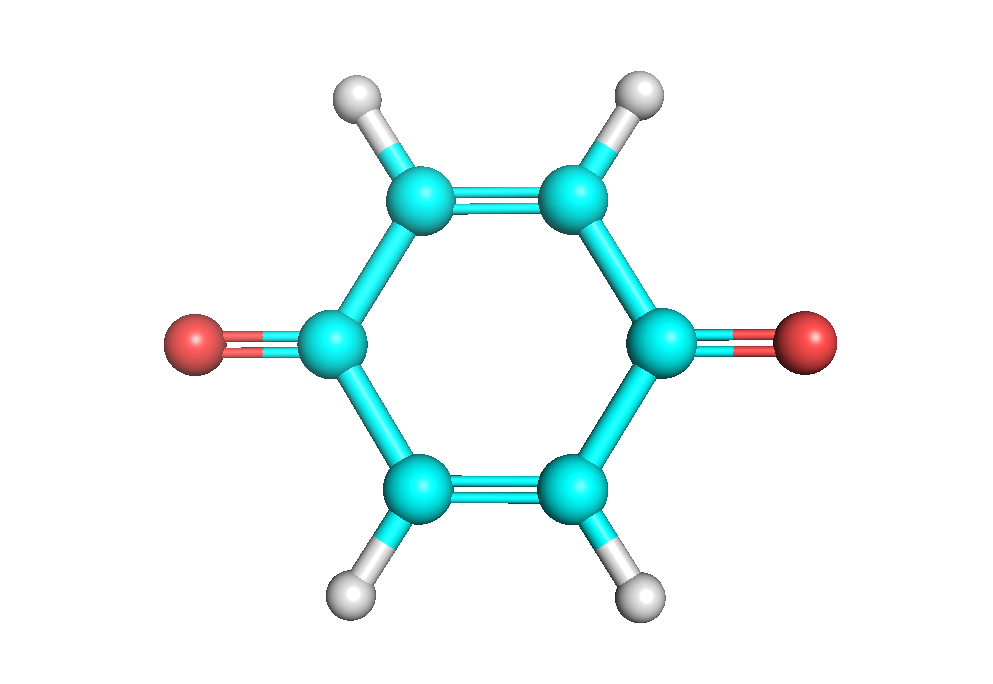
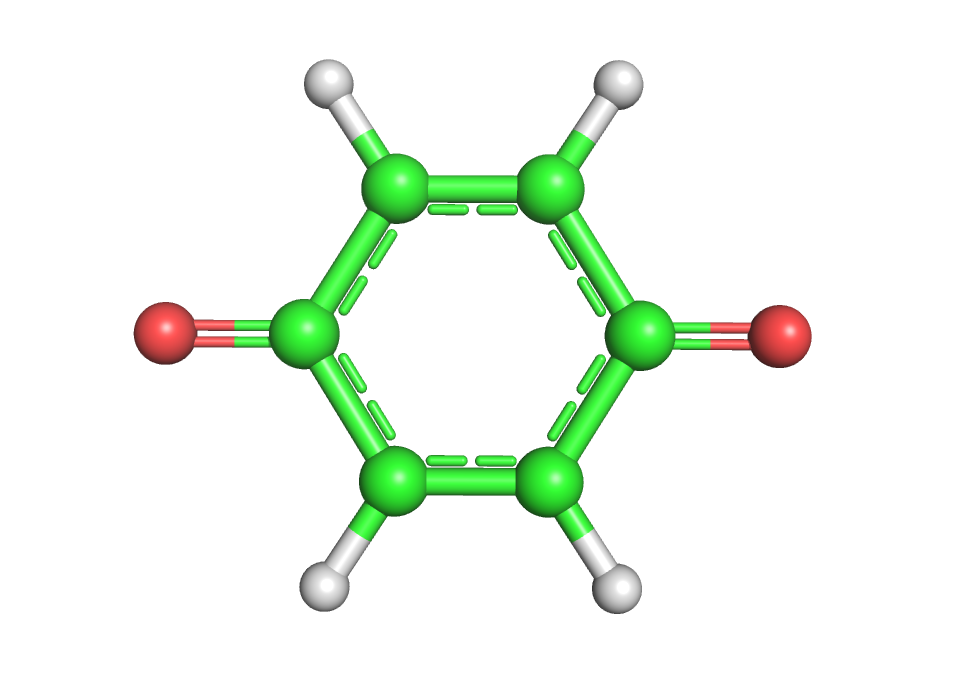
Рис. 5. Структура, полученная obgen. Рис. 6. Структура, полученная Mopac.
В целом, полученные структуры схожи и обе характеризуются плоской геометрией, но представление связей различно.
Далее определялась геометрия дианиона молекулы. В первую строчку mop файла было добавлено слово CHARGE=-2, атомы кислорода были указаны как несущие отрицательный заряд:
... C 3.09700 1 -1.26700 1 -0.12500 1 O(-) 1.67000 1 -0.00400 1 -1.53600 1 O(-) 5.45700 1 0.02000 1 2.22300 1 H 2.66300 1 2.19000 1 -0.55900 1 ...
Полученный mop файл был подан на вход Mopac, результат его выдачи был переведен в pdb c помощью babel и проанализирован в Pymol. Полученная структура также плоская и похожа на предыдущие. На рис. 7 и 8 отмечены отличия длин связей в полученной структуре дианиона (рис. 8) и структуре хинона (рис. 7).
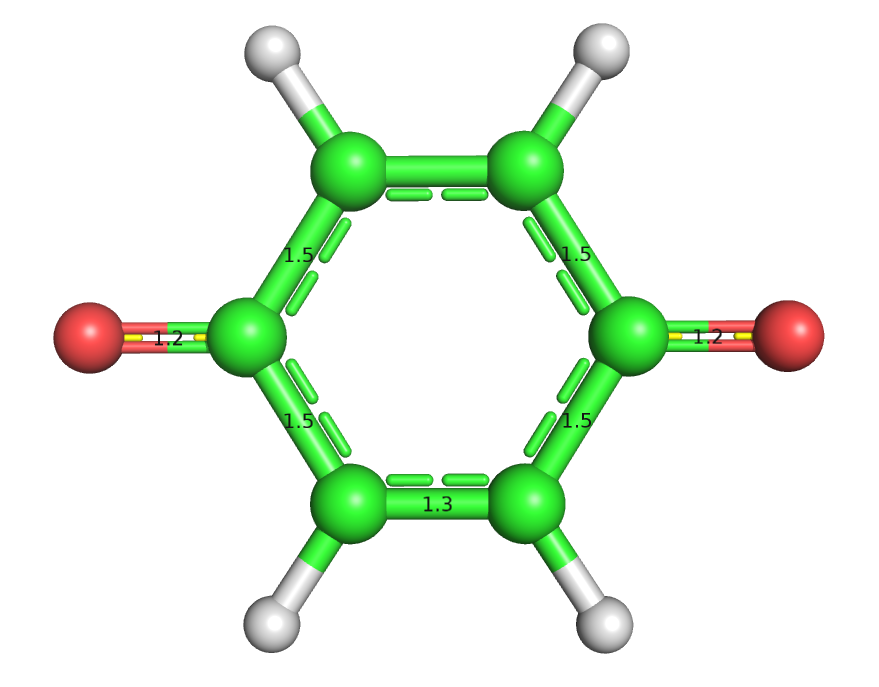
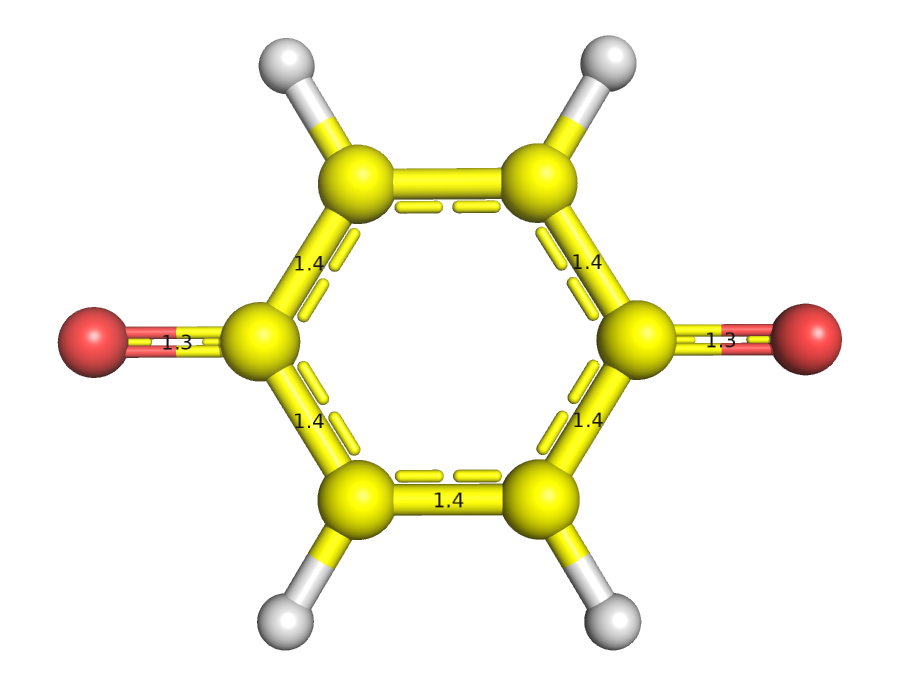
Рис. 7. Структура хинона. Рис. 8. Структура его дианиона.
4. Тиминовый димер
Известно, что ультрафиолет может превращать тимины в тиминовые димеры, так же известно, что ДНК фотолиаза при облучении ультрафиолетом востановливает основания тиминов до нормального. Чтобы увидеть переход из димера в тимины при возбуждении системы, сымитируем возбуждение, ионизируя оба кольца, т.е. указывая заряд системы +2, и снова оптимизируем полученое возбуждённое состояние при заряде 0.
Проводим оптимизацию структуры димера в Mopac как в предыдущих пунктах: babel -ipdb td.pdb -omop td_opt_n.mop -xk "PM6"; MOPAC2009.exe td_opt_n.mop; babel -imopout td_opt_n.out -opdb td_opt_n.pdb. После первой оптимищации при заряде системы 0 была получена сходная структура, сравнение которой с исходной представлено на рис. 9.
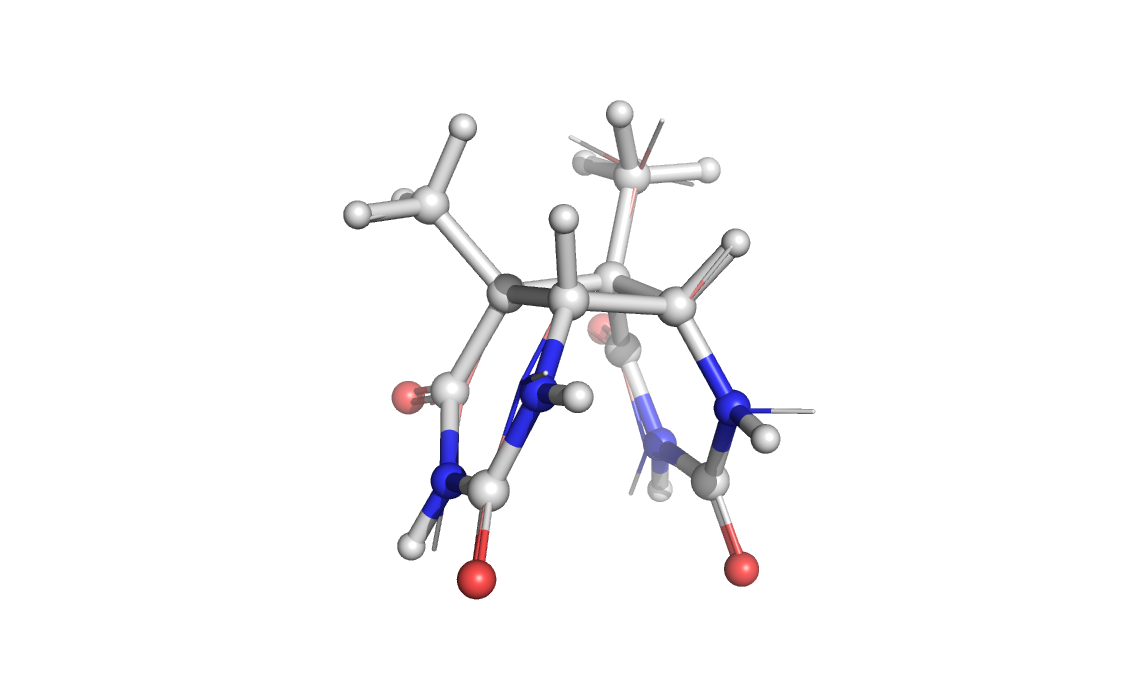
Рис. 9. Наложение оптимизированной структуры (ball and sticks) на исходную (lines)
Далее имитируем возбуждение молекулы. В результате кольца расходятся, и пропадает одна из связей между димерами. Сравнение полученной структуры с предыдущей - на рис. 10.
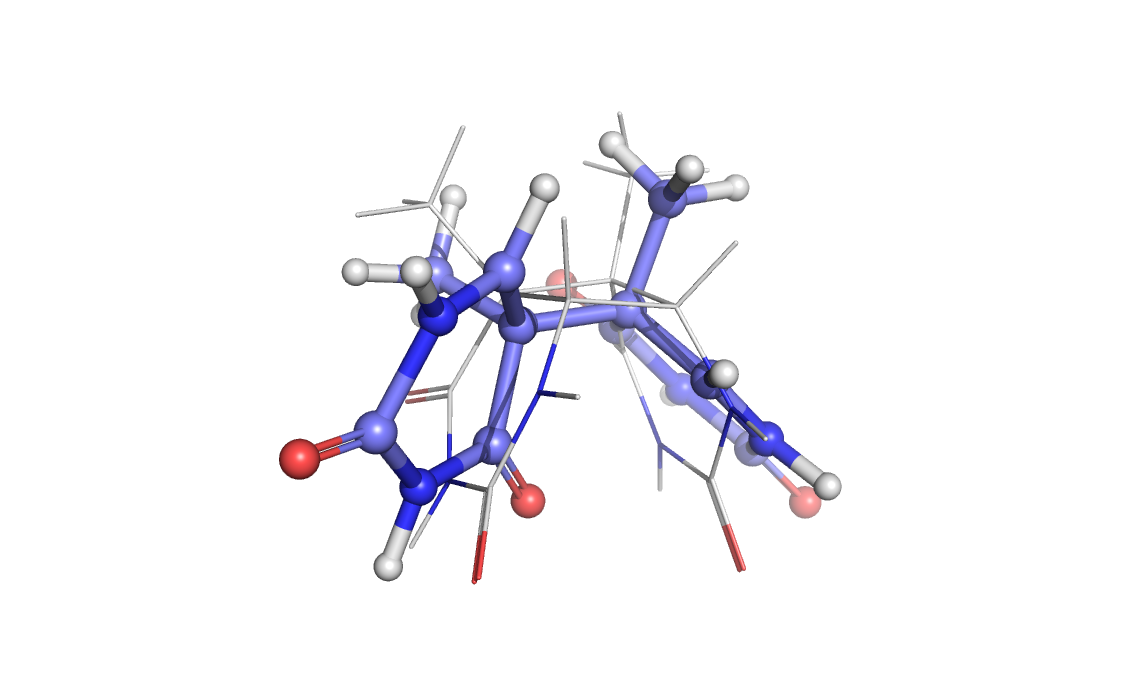
Рис. 10. Наложение оптимизированной при заряде системы +2 структуры (ball and sticks) на оптимизированную при заряде 0 (lines)
Снова проводим оптимизацию незаряженной системы. Исчезает вторая связь между димерами, тимины отдаляются друг от друга.
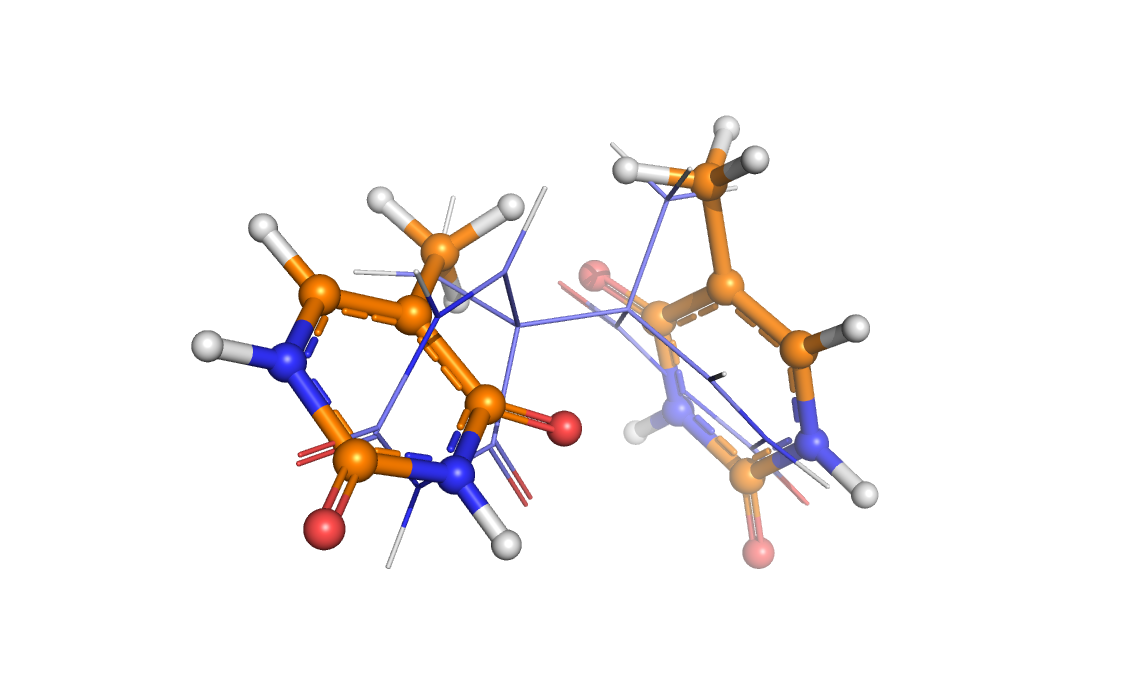
Рис. 11. Наложение оптимизированной при заряде системы 0 структуры (ball and sticks) на оптимизированную при заряде +2 (lines)
Итак, распад димера произошел в два этапа. Сравнение энергий (Эв) для состояний: -3273.58217 (димер), -3253.90834 (переход) и -3273.69661 (мономеры), показывает, что существование тиминов как мономеров немного выгоднее, чем в виде димера, что, вероятно, повлияло на то, что система не перешла в исходное состояние в заданных условиях.
© Eugenia Prokhorova, Евгения Прохорова, 2014