Докинг низкомолекулярных лигандов в структуру белка
1-4.
В банке pdb найдена SMILES нотация для NAG на странице структуры 1lmp. Нотация сохранена в файл nag.smi. C помощью obgen построена 3D структура этого сахара в pdb формате.
Скриптом prepare_ligand4.py из пакета Autodock tools создан pdbqt файл лиганда:
Так же, скриптом prepare_receptor4.py из пакета Autodock tools создан pdbqt файл белка, построенной на прошлом занятии модели 5 (взята как наилучшая):
5-10.
Создан файл с параметрами докинга vina.cfg. Область структуры белка, в которой будет происходить поиск места для связывания, задана как куб с неким центром. Координаты центра определены из модели 5, построенной на прошлом занятии. Центр масс определен с помощью PyMol, команда pseudoatom, однако оказался он вдали от лиганда (рис. 1, серая сфера). Поэтому было решеновзять за центр массы координаты одного из атомов лиганда. Итак, построен файл vina.cfg с заданным содержанием.
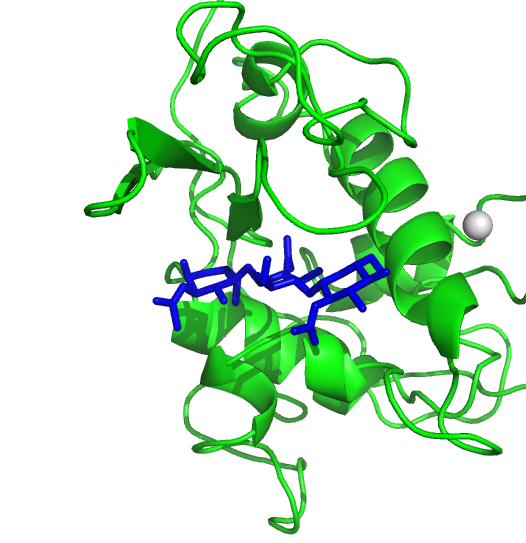
Рис. 1.
Первый докинг:
Просмотрен полученный файл nag_prot.log. Энергии трех лучших расположений и геометрические разницы между ними:
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -4.8 0.000 0.000
2 -4.6 1.948 3.739
3 -4.5 1.382 2.695
Файлы nag_prot.pdbqt и prot.pdbqt загружены в PyMol. На рис. 2 отображены все состояния с помощью:
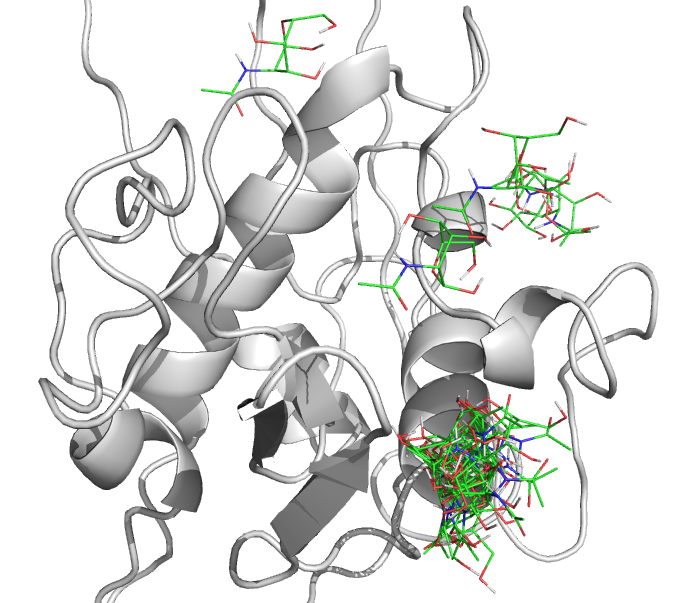
Рис. 2.
На рис. 2 видно, что в большинстве случаев, в том числе в трех случаях с наилучшей энергией, NAG связался в нижнем сайте.
Далее проведен докинг, рассматривая подвижность некоторых боковых радикалов белка. Белок был разбит на две части - подвижную и неподвижную. Для подвижной части выбраны 3 из аминокислот, использованных в прошлом задании для позиционирования лиганда и затем проведен докинг:
Просмотрен полученный файл nag_prot_flex.log. Энергии трех лучших расположений и геометрические разницы между ними:
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -4.7 0.000 0.000
2 -4.6 1.499 2.904
3 -4.5 1.117 2.191
В PyMol загружены файлы nag_prot_flex.pdbqt и prot_rigid.pdbqt. На рис. 3 отображены все состояния.
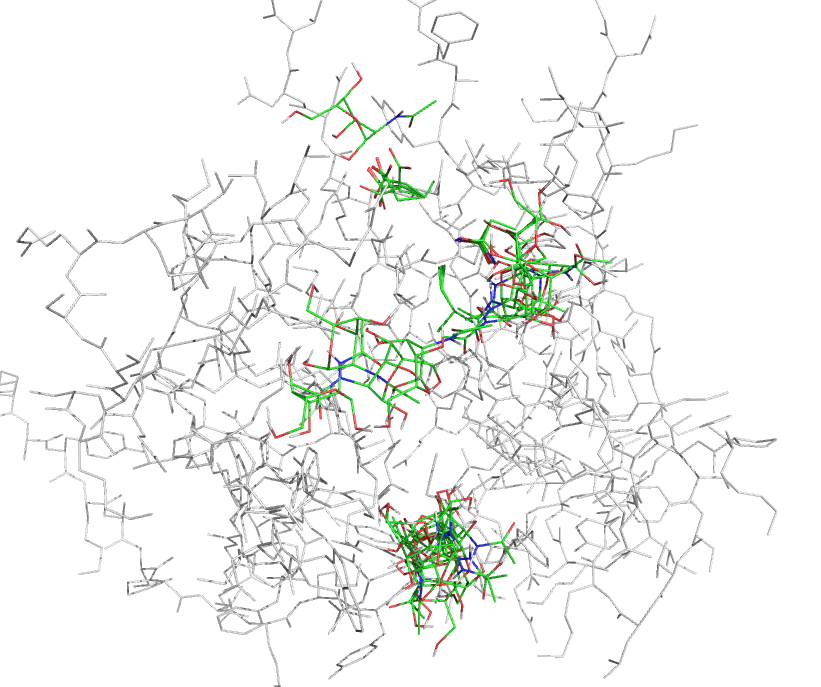
Рис. 3.
По сравнению с обычным докингом, связывание в нижнем сайте оказалось менее частым, видимо, изменения положений остатков ухудшали энергию связывания лиганда в нем. Полученные значения энергий связывания лиганда схожи, наилучшая энергия даже хуже, при этом алгоритм работал дольше.
11-12.
Созданы 4 лиганда, где метильный радикал заменён. Для каждого из этих лигандов проведен докинг. Просмотрены полученные варианты связывания nagoh_prot.pdbqt, nagnh2_prot.pdbqt, nagh_prot.pdbqt, nagph_prot.pdbqt. Наилучшие по энергии связывания снова наблюдаются в нижнем сайте. Результаты представлены в виде таблицы из трех лучших энергий для каждого лиганда:
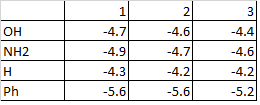
Далее проведен докинг с подвижными радикалами для трех лигандов (кроме Н). Просмотрены полученные варианты связывания nagoh_prot_flex.pdbqt, nagnh2_prot_flex.pdbqt, nagph_prot_flex.pdbqt. Результаты представлены в виде таблицы из трех лучших энергий для каждого лиганда:
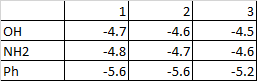
Видно, что значения энергии практически не изменились, но немного ухудшились. При этом увеличилось время работы. Изменение визуального расположения лигандов схоже с таковым для случая без замены радикалов в NAG.
© Eugenia Prokhorova, Евгения Прохорова, 2014