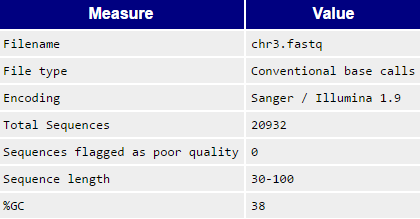
Чтения экзома, картирующиеся на участок хромосомы человека. Файлы с одноконцевыми чтениями в формате fastq: chr3.fastq
Хромосома человеческого генома (сборка версии hg19): chr3.fasta
Для анализа "сырых" прочтений можно применить программу FastQC. Она проводит статистические анализы, результаты которых можно использоать для оценки качества данных и выявления проблемм в прочтении последовательности. Нужно заметить, что в выданных нам чтениях адаптеры уже удалены.
Данная программа установлена на kodomo, для её вызова использовалась команда fastqc chr2.fastq
Результат - архив (.zip), из который содержит отчет о программе в виде html файла: fastqc_report.html
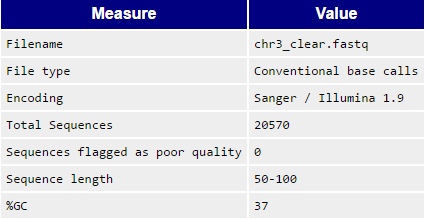
Очистку чтений производится с помощью программы Trimmomatic. Она также установлена на kodomo.
Для вызова программы Trimmomatic использовалась команда java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr3.fastq chr3_clear.fastq TRAILING:20 MINLEN:50
Необходимо отсечь с конца чтения нуклеотиды с неудовлетворительным качеством (< 20), для этого был указан параметр TRAILING:20. Также требовалось оставить только прочтения длины не менее 50, поэтому был указан параметр MINLEN:50.
Результат (после анализа через FastQC): fastqc_clear_report.html
Сравнение качества до (картинки слева) и после (картинки справа) чистки:
Число ридов уменьшилось на 326, на 1% уменьшилось содержание GC пар.
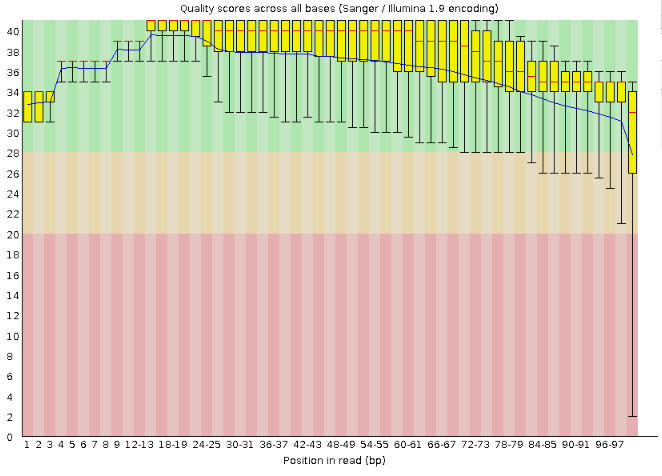
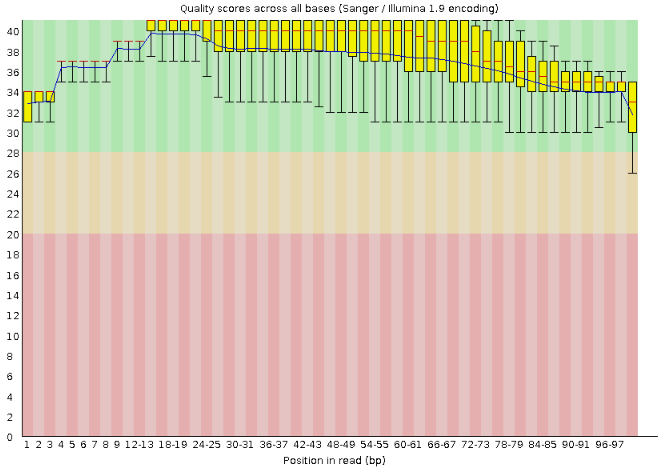
После чистки значительно число усов, выходящих из зеленой области. Медиана в обоих случаях практически не изменилась, математическое ожидание повысилось. Все это говорит о более высоком качестве оставшихся чтений. Таким образом, Trimmomatic вырезал низкокачественные чтения и оставил только те, с которыми можно нормально работать.
Для откартирования чтений применяется программа BWA, которая установлена на kodomo. Анализ осуществляется с помощью программы Samtools также установленной на kodomo.
Для построения и анализа выравнивания были выполнены следующие шаги:
1) Индексирование референсной последовательности, команда bwa index chr3.fasta
2) Построение выравниваения очищенных чтений с референсной последовательностью, команда bwa mem chr3.fasta chr3_clear.fastq > chr3.sam
3) Выравнивание необходимо перевести в бинарный формат, команда samtools view -b chr3.sam > chr3.bam
4) Теперь отсортируем выравнивание по координате в референсе начала чтения, команда samtools sort chr3.bam -T pr -o chr3_sort.bam
5) Проведем индексирование выравнивания, команда samtools index chr3_sort.bam
6) А теперь получим количество картированных чтений samtools idxstats chr3_sort.bam > reads3.out
Результат работы - reads3.out
На хромосому были откартированы 20569 чтений, 1 чтение откартировано не было (цифра 3 появляется из-за учета последней строки, что можно считать ошибкой программы при интерпретации данных.
Создание файла с полиморфизмами в формате .bcf осуществляется командой samtools mpileup -uf chr3.fasta chr3_sort.bam -o pol.bcf
Далее необходимо создать файл со списком отличий между референсом и чтениями в формате .vcf. Для этого можно использовать команду bcftools call -cv pol.bcf -o pol.vcf
Результат - pol.vcf
Всего было найдено 235 различных полиморфизмов, большая часть которых - замены, но также есть и делецииЮ, и вставки.
| Информация о трёх полиморфизмах | |||||
|---|---|---|---|---|---|
| Координата | Тип | В референсе | В прочтении | Качество прочтения | Глубина прочтения |
| 41291081 | Замена | G | A | 221.999 | 25 |
| 41841811 | Вставка | TATTA | TATTAATTA | 217.468 | 29 |
| 41935127 | Делеция | CATTAT | CAT | 217.486 | 115 |
Для аннотации полученных SNP используется программа ANNOVAR.
Файл надо подготовить к аннотации. Для этого удалим из .vcf все индели (вручную), получим файл pol_naindel.vcf. Затем применим скрипт convert2annovar.pl.
Для скрипт запускается командой perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 pol_noindel.vcf -outfile pol.avinput
Результат - pol.avinput
При помощи скрипта annotate_variation.pl можно провести 3 типа аннотации по базам данных, основанных на: генной разметке (gene-based annotation); разметке других регионов генома (region-based annotation); фильтрации (filter-based annotation).
1)Аннотация по базе refgene:
Используемая команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -out chr3.refgene -build hg19 pol.avinput /nfs/srv/databases/annovar/humandb
Полученные файлы:
Категории, указывающие на расположнение плиморфизмов (1 колонка файла chr3.refgene.variant_function):
Всего файл содержит информацию о 218 полиморфизмах (замен и делеций в нем нет), из них в экзонах находятся 13, в 5-нетранслируемых областях 6, остальные 199 расположены в интронах.
Большая часть полиморфизмов расположена в интронах, то есть эти изменения не проявляются при транслации. Это ожидаемо, так как мутации в экзнах могут привести к испорченному белку и зачастую к летальному исходу или к появлению наследственного заболевания. На мутации в нетранслируемых областях не действует отбор, поэтому они накапливаются в геноме.
Найденные полиморфизмы находятся в следующих генах: ULK4 и FNDC3B.
Ген ULK4 кодирует серин/треонин киназу (СТК). Эти ферменты образуют целое семейство, члены которого играют важную роль в процессах роста нейронов и эндоцитозе. Кодируемый белок, вероятно, участвует в удлинении и миграции нейронов. Исследования указывают на связь вариаций в этом гене с кровяным давлением и артериальной гипертензией. Отдельные вариации в этом гене могут также быть связаны с психическими расстройствами, включая шизофрению и биполярное расстройство (маниакально-депрессивный психоз). В гене обнаружено 10 замен, 8 из которых несинонимичны, т.е. произошла замена аминокислоты в белковой последовательности.
Ген FNDC3B кодирует белок 3B, содержащий домен фибронектина III типа (может быть регулятором адипогенеза).В гене обнаружено 3 замены, 2 из которых являются синонимичными (замены аминокислоты не произошло).
2)Аннотация по базе GWAS (клиническая картина):
Используемая команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -out chr3.gwas -build hg19 -dbtype gwasCatalog pol.avinput /nfs/srv/databases/annovar/humandb/
Полученные файлы:
Интерпретация результатов: описывается 4 замены. Замена в гене ULK4 связана с кровяным давлением. Полиморфизм в гене GNL3 влияет на уровень гормона адипонектина, который участвует в регуляции уровня глюкозы и расщепления жирных кислот. Обе замены в гене FNDC3B влияют на рост.
3)Аннотация по базе 1000 genomes:
Используемая команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr3.dbsnp -build hg19 -dbtype snp138 pol.avinput /nfs/srv/databases/annovar/humandb/
Результат: 3 файла, один с полиморфизмами, имеющими rs в 1000 genomes; второй с полиморфизмами, не имеющими rs в 1000 genomes, третий с отчетом о работе программы.
Частота snp представлена в файле 1000g.hg19_ALL.sites.2014_10_dropped
Как видно из данных частота полиморфизмов имеет довольно большой разброс: наименьшая частота полиморфизма 0.000599042, наибольшая - 0.996805
4)Аннотация по базе Dbsnp:
Используемая команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr11.dbsnp -build hg19 -dbtype snp138 pol.avinput /nfs/srv/databases/annovar/humandb/
Полученные файлы:
В первом файле представлены snp, имеющие snp. Из найденных snp rs имеют 179 штук. Во втором файле - snp, не имеющие rs (39 штук).
5)Аннотация по базе Clinvar:
Используемая команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr3.clincar -buildver hg19 -dbtype clinvar_20150629 pol.avinput /nfs/srv/databases/annovar/humandb/
Полученные файлы:
В Clinvar не оказалось анонсировано ни одного snp из найденных мной на предложенной хромосоме.
Файл с результатами аннотирования: all.xlsx
Файл со сводной таблицей: annotate.xlsx
© Борисов Евгений 2016