Практикумы 10 и 11
Сравнение состава систем рестрикции-модификации, закодированных в двух штаммах одного вида
Выданный организм: Enterococcus faeciumРеференсный геном в формате fasta (gzipped) Выходной файл сервиса Calculate contrast В референсном геноме этой бактерии не обнаружено недопредставленных сайтов (с порогом 0.78) рестрикции-модификации.
Геном из метагенома кишечника человека в формате fasta (gzipped) В геноме бактерии из кишечника обнаружен 1 недопредставленный сайт (соотношение Карлина - 0.769) рестрикции-модификации - GGCGCC. Однако вряд ли он позволяет говорить о наличии Р-М системы, потому что мало наблюдений: в геноме 30 сайтов, а алгоритм Карлина предсказывал 38.99. Есть ещё 2 сайта, соотношение Карлина для которых не превышает 0.79, но для них наблюдений ещё меньше.
Выходной файл сервиса Calculate contrast Полученные сведения не позволяют сделать сколько-нибудь определённый вывод. Возможно, иммунитет этой бактерии в большей степени определяется другими системами.
Последовательности Шайн-Дальгарно в геноме Elusimicrobium minutum
Геном моей бактерии Elusimicrobium minutum по данным NCBI Genome содержит 1519 генов. К сожалению, они очень плохо аннотированы: только для 4 из них предсказаны продукты (1 из них - hypothetical protein), поэтому "хорошая" выборка формировалась с использованием критерия длины (брались гены длиной более 1000 п.н.). С помощью предлагаемого скриптов features2CDSs.py и fragments2fasta.py я вырезал участки перед стартами трансляции "хороших" генов длиной в 16 п.н. Ссылка на fasta-файл с участками перед стартами трансляции для "хороших" генов Для поиска мотива использовалась программа meme на сервере kodomo.Команда: meme SD.fasta -dna -nmotifs 1 -minw 5 -maxw 7 -o mem
-dna означает, что используется алфавит ДНК, -nmotifs определяет, сколько мотивов искать, -minw определяет минимальную длину мотива, а -maxw - максимальную, -o используется для задания директории с выходными файлами.
Рис. 1. Лого полученного мотива.

Рис. 2. Позиционно-весовая матрица для полученного мотива.
Затем были вырезаны участки перед стартами трансляции длиной 21 п.н. для всех генов и с помощью программы fimo на сервере kodomo с использованием полученной ранее позиционно-весовой матрицы было проверено наличие последовательности Шайна-Дальгарно в этих участках.
Команда: fimo --output-pthresh 1e-2 ./mem/meme.txt SDall.fasta
Опция --output-pthresh позволяет установить порог p-value, выше которого находки не рассматриваются. ./mem/meme.txt содержит позиционно-весовую матрицу, SDall.fasta - файл с участками перед стартами трансляции.
Ссылка на файл xls с генами, в которых нашлась последовательность Шайна-Дальгарно
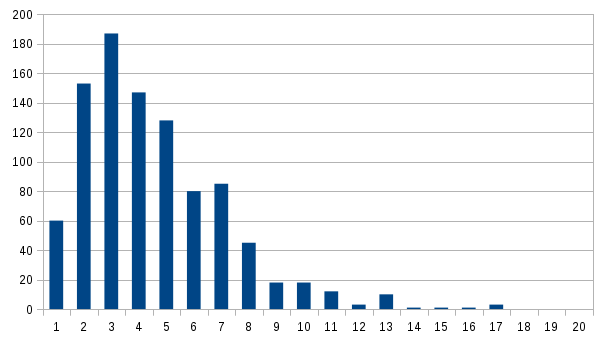
Рис. 3. Гистограмма расстояний найденных последовательностей ШД от стартов трансляции
Рис. 4. Лого всех найденных мотивов
Процент генов, перед стартами трансляции которых была найдена последовательность Шайна-Дальгарно составил 66,16 (1006 генов их 1519). К сожалению, последовательности Шайна-Дальгарно в геноме моей бактерии пока никого не заинтересовали, поэтому мне не удалось найти вообще никакой литературы о их наличии (сам организм упоминается только в двух статьях при поиске в Pubmed). Найденный мотив, всё же, похож на последовательность Шайна-Дальгарно, встречающуюся в геноме Escherichia coli, поэтому можно говорить о её наличии в этом организме. Однако, как было указано выше, она есть далеко не у всех генов.
Сайты связывания транскрипционного фактора
Контроль качества с помощью Fastqc показал, что с данными можно работать, дополнительная обработка с помощью Trimmomatic не требуется. Однако в некоторых ячейках Illumina качество было плохим, что отражено на рис. 2.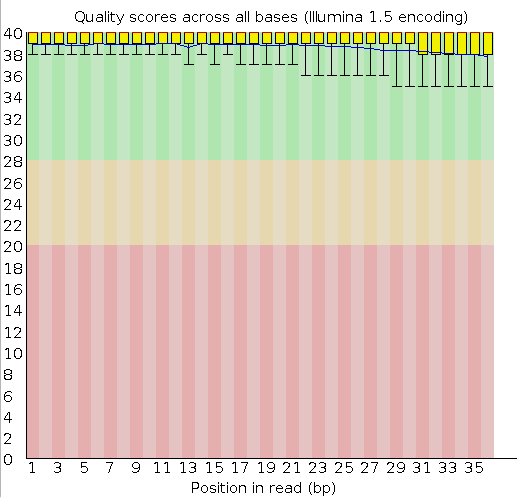
Рис. 5. Качество позиций в ридах.
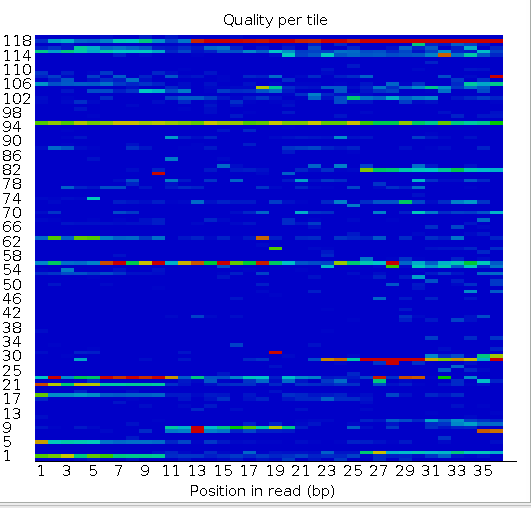
Рис. 6. Относительное качество секвенирования по ячейкам.
Таблица 1. Использованные команды
| Команда | Значение |
| bwa mem /srv/databases/ngs/hg19/GRCh37.p13.genome.fa chipseq_chunk38.fastq > chipseq_chunk38.sam | Картирование ридов из файла chipseq_chunk38.fastq, выдача записана в файл chipseq_chunk38.sam. |
| samtools view -bo chipseq_chunk38.bam chipseq_chunk38.sam | Конвертация файла в формате sam в бинарный файл bam. Опция -b - конвертировать в bam, -o - после этой опции нужно писать выходной файл. Опция -S устарела, применялась в ранних версиях, если входной файл был в формате sam. |
| samtools sort chipseq_chunk38.bam -T chip_temp -o chipseq_chunk38.sorted.bam | Сортировка bam-файла с использованием временного файла chip_temp и выходного chipseq_chunk38.sorted.bam. |
| samtools index chipseq_chunk38.sorted.bam | Индексирование сортированного файла.s |
| samtools idxstats chipseq_chunk38.sorted.bam > chipseq_chunk38.idxstats | Количество картированных и некартированных ридов по хромосомам и контигам. |
| samtools view -c chipseq_chunk38.sorted.bam | Выдаёт количество картированных ридов. |
| macs2 callpeak -t chipseq_chunk38.sorted.bam --nomodel | Получение файлов с пиками |
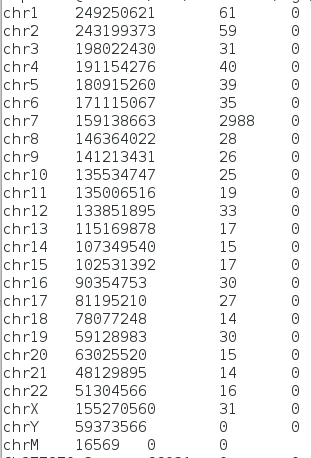
Рис. 7. Часть файла chipseq_chunk38.idxstats с количеством ридов по хромосомам. Из этого заключаю, что этот кусок ChIP-seq принадлежал 7 хромосоме.
С помощью программы MACS удалось получить два пика на 7 хромосоме. Ширина первого (слева направо) пика - 207 п.н., второго - 485 п.н. Пики лежат вне генов, скоры - 7.87 и 15.11 соответственно. В файле .narrowPeak есть ещё один пик - на контиге JH159134.2.

Рис. 8. Визуализация файла .narrowPeak в UCSC Genome Browser. Красным помечены пики.
Поиск сигналов TATA-бокс связывающего белка (TBP) в геноме человека
Таблица 1 Сайты связывания TBP в геноме человека| Название | Хромосома, позиция, ориентация гена | Длина гена | Изображение профиля TBP в мелком масштабе | Изображение профиля TBP в крупном масштабе | Координата TATA-бокса относительно старта транскрипции | Обсуждение |
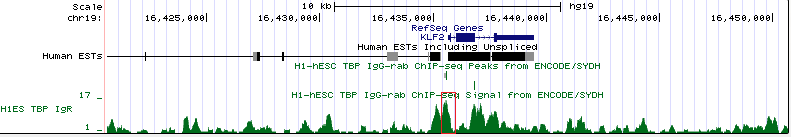
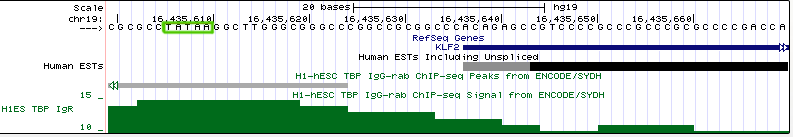
| KLF2 - Kruppel-like factor 2 | chr19:16,435,637, прямая цепь | 3,837 п.н. |
|
|
-22 | Пик хотя и отчётливый, но двойной, поэтому не совсем понятно, где же на самом деле садится на ДНК TBP. Это обстоятельство усугубляется тем, что пик не намного превосходит шум. |
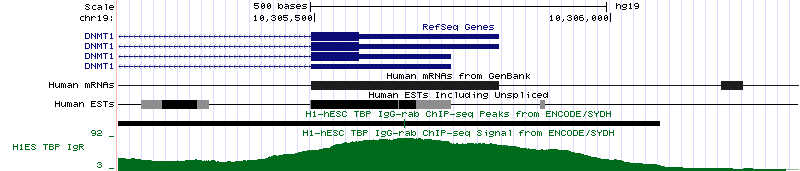
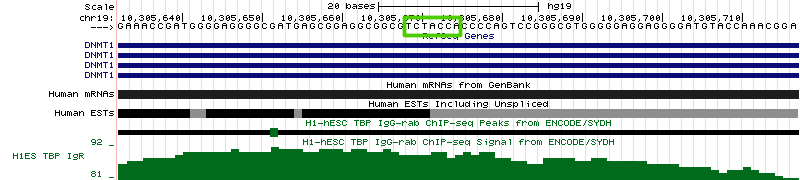
| DNMT1 | chr19:10,305,755, обратная цепь | 61,734 п.н. |
|
|
-107 | Пик не очень сильно отличается от уровня шума по геному, поэтому не совсем понятно, реален ли он. |
| FOSB | chr19:45,971,253, прямая цепь | 7185 п.н. |

|
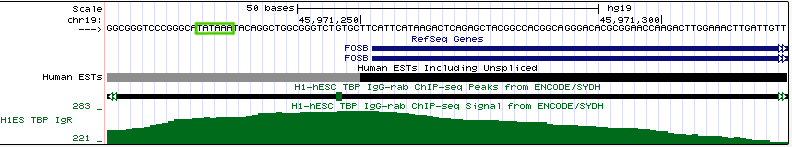
|
-31 | Пик хотя и слегка расплывчатый, но довольно сильно превосходит уровень шума, что позволяет предполагать наличие сайта посадки TBP перед этим геном. |
| ATP2B2 | chr3:10,547,268, обратная цепь | 181,562 п.н. |
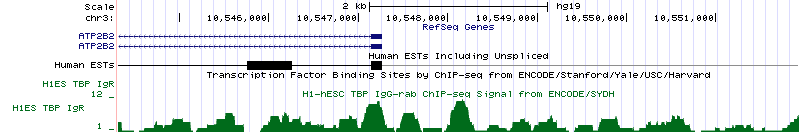
|
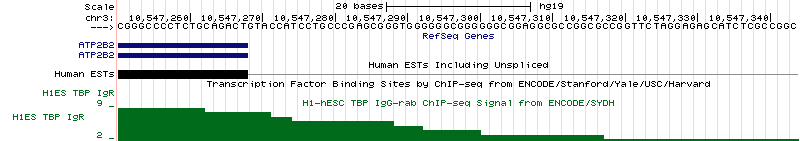
|
Нет | По сравнению с шумом, пика нет, как нет и сайта связывания TBP. |