1Сравнение выравниваний одних и тех же последовательностей разными программами
Для построения выравниваний были использованы последовательности из семейства белковых доменов PF00167 (Fibroblast growth factor).
Гиперссылки на сравниваемые выравнивания в формате fasta:
Для того, чтобы сравнить выравнивания, я использовала код, написанный моими однокурсниками.
MUSCLE & MAFFT
| Muscle | Mafft | Длина |
|---|---|---|
| (1, 9) | (1, 9) | 2 |
| (11,12) | (11,12) | 4 |
| (20,24) | (20,24) | 5 |
| (28,37) | (28,37) | 10 |
| (43,81) | (44,82) | 39 |
| (95,101) | (94,100) | 7 |
| (106,112) | (105,111) | 7 |
| (125,129) | (124,128) | 5 |
| (131,134) | (130,133) | 4 |
| (140,171) | (139,170) | 32 |
(40,41)
(120,119)
Длина второго выравнивания: 170
Количество одинаково выровненных колонок: 126
Процент одинаково выровненных колонок от длины первого выравнивания: 73.68%
Процент одинаково выровненных колонок от длины второго выравнивания: 74.12%
MUSCLE & TCOFFEE
| Muscle | Tcoffee | Длина |
|---|---|---|
| (1,9) | (1,9) | 9 |
| (11,12) | (11,12) | 2 |
| (14,17) | (14,17) | 4 |
| (20,24) | (20,24) | 5 |
| (27,28) | (27,28) | 2 |
| (31,35) | (31,35) | 5 |
| (43,53) | (44,54) | 11 |
| (59,82) | (61,84) | 24 |
| (95,101) | (98,104) | 7 |
| (107,115) | (110,118) | 9 |
| (126,127) | (129,130) | 2 |
| (131,135) | (134,138) | 5 |
| (140,154) | (143,157) | 15 |
| (159,162) | (164,167) | 4 |
(40,41)
(57,59)
(129,132)
Длина второго выравнивания: 179
Количество одинаково выровненных колонок: 107
Процент одинаково выровненных колонок от длины первого выравнивания: 62.57%
Процент одинаково выровненных колонок от длины второго выравнивания: 59.78%
Выравнивание MAFFT оказалось ближе к выравниванию MUSCLE: у этой пары нашлось больше совпадающих колонок, чем у пары MUSCLE–TCoffee, и доля одинаково выровненных позиций относительно длины выравнивания тоже выше. В паре MUSCLE–TCoffee количество совпадающих блоков больше (14 против 10 у MUSCLE–MAFFT), однако часть этих блоков короче, чем в случае MUSCLE–MAFFT, а само выравнивание TCoffee получилось самым длинным за счёт большего числа гэпов.
2Сравнение выравнивания по совмещению структур с выравниванием MSA
Для построения выравниваний были использованы последовательности из семейства белковых доменов PF00167 (Fibroblast growth factor): 1axm, 1bar, 1bfb. Выравнивание по совмещению структур было получено программой PDBeFold.
Ссылки на выравнивания в формате fasta:
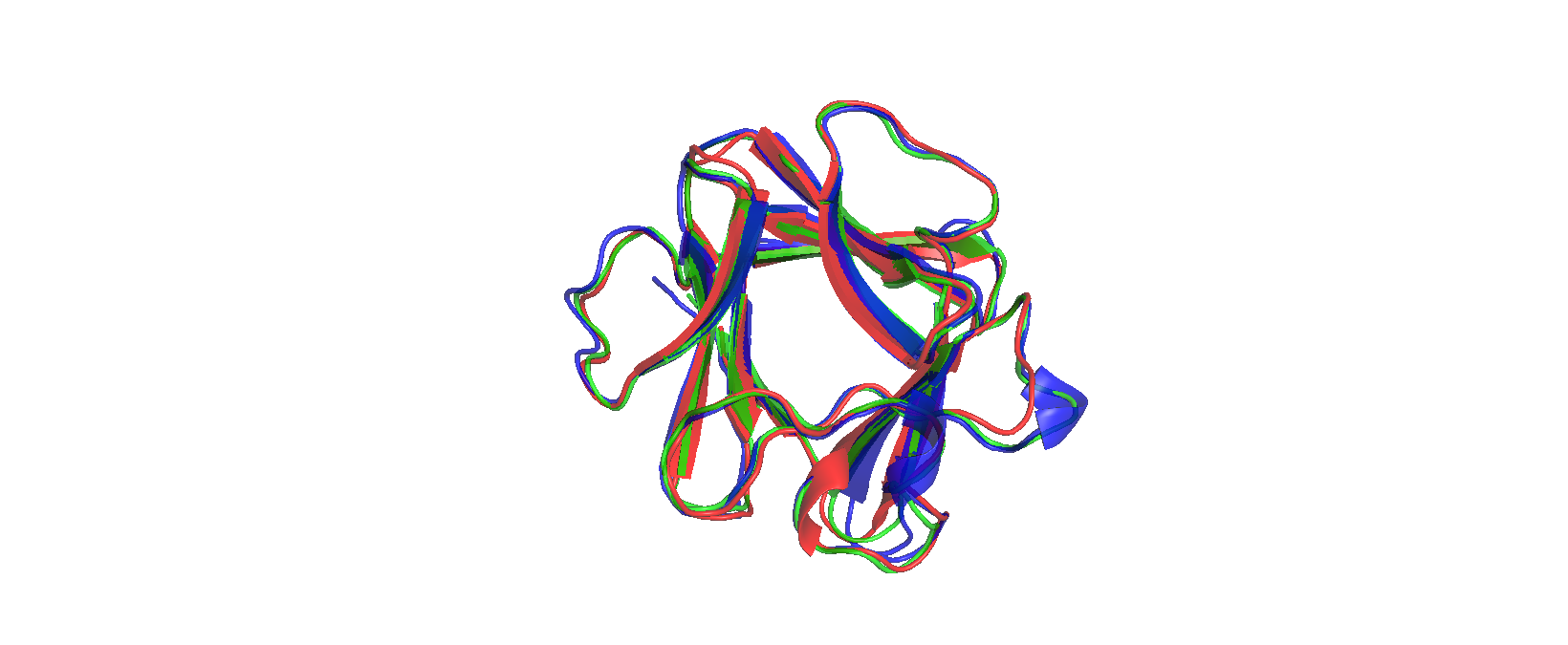
При структурном выравнивании трёх белков их центральные участки накладываются очень хорошо, а основные элементы вторичной структуры (α‑спирали и β‑листы) занимают сопоставимые позиции во всех трёх цепях. Петли и концевые участки демонстрируют больший разброс по положению в пространстве, что соответствует пониженной консервации в этих регионах в выравнивании.
В JalView аннотация secondary structure показывает, что спирали попадают в протяжённые хорошо выровненные блоки с высокой conservation и quality, тогда как в участках без регулярной вторичной структуры наблюдается больше гэпов и вариабельности. Это говорит о том, что структурное выравнивание корректно фиксирует консервативное структурное ядро домена, а различия между белками в основном приходятся на гибкие петли и вставки.
| PDBeFold | Muscle | Длина |
|---|---|---|
| (1,95) | (1,95) | 95 |
| (99,130) | (99,130) | 32 |
Процент одинаково выровненных колонок от длины первого выравнивания: 98.46%
Процент одинаково выровненных колонок от длины второго выравнивания: 98.46%
При сравнении выравнивания, полученного из совмещения структур, с выравниванием MUSCLE видно, что в области структурного ядра домена оба метода дают практически одинаковый результат: основные элементы вторичной структуры (α‑спирали и β‑листы), которые хорошо накладываются в 3D, попадают в одни и те же протяжённые блоки с высокой conservation и quality. Степень согласованности двух выравниваний количественно тоже очень высокая: около 98 % колонок совпадают полностью, а различия в расположении гэпов наблюдаются лишь в одном локальном участке, вероятно соответствующем гибкой петле или вставке. Таким образом, MUSCLE практически полностью воспроизводит структурное выравнивание для выбранных белков и заметно расходится с ним только там, где сама структура наиболее вариабельна, тогда как в консервативном структурном ядре соответствие между методами практически полное.
4Краткое описание MUSCLE
MUSCLE (MUltiple Sequence Comparison by Log‑Expectation) — программа для множественного выравнивания белков и нуклеиновых кислот, предложенная R. Edgar в 2004 году. Она сочетает прогрессивное выравнивание с быстрыми эвристиками и итеративным улучшением, что обеспечивает одновременно высокую точность и эффективность при работе с большими наборами последовательностей. [1]
Алгоритм MUSCLE включает три основные стадии. Сначала быстро оцениваются расстояния между последовательностями (на основе k‑меров) и строится первоначальное дерево, по которому выполняется черновое прогрессивное выравнивание. Затем по уже выровненным последовательностям пересчитываются расстояния, строится улучшенное дерево и проводится второе, более точное прогрессивное выравнивание. На заключительном этапе выполняется итеративный refinement: дерево разрезают по рёбрам, заново выравнивают две группы последовательностей и принимают новый вариант, если он улучшает целевую функцию (log‑expectation score, LE‑score). [1]
Ключевые преимущества MUSCLE: (1) высокая точность на стандартных тестовых наборах (BAliBASE, SABmark, SMART, PREFAB) — качество выравнивания сопоставимо или лучше, чем у ClustalW и T‑Coffee; (2) высокая скорость и умеренное потребление памяти, особенно в режиме fast, который даёт точность, близкую к ClustalW, но на 2–3 порядка быстрее на больших наборах последовательностей. Основные ограничения — снижение точности на очень сильно дивергентных последовательностях и отсутствие явного учёта структурной информации (3D‑структуры, экспериментальной вторичной структуры), поскольку алгоритм использует только данные о последовательности и статистические оценки профилей. [1]
[1] MUSCLE: a multiple sequence alignment method with reduced time and space complexity
Robert C Edgar
doi:10.1186/1471-2105-5-113