Программы множественного выравнивания
Различные программы множественного выравнивания
Построим множественные выравнивания программами MUSCLE, MAFFT, CLUSTAlW для последовательностей белков UVRA следующих видов:
Streptococcus pyogenes serotype M3 (ID: UVRA_STRPQ, AC: P0CZ41), Streptococcus pneumoniae serotype 4 (ID: UVRA_STRPN AC: P63384),
Streptococcus mutans serotype c (ID: UVRA_STRMU AC: P72481), Staphylococcus aureus (ID: UVRA_STAAR AC: Q6GIN2),
Escherichia coli (ID: UVRA_ECOLI AC: P0A698), Bacillus subtilis (ID: UVRA_BACSU AC: O34863),
Neisseria gonorrhoeae (ID: UVRA_NEIGO AC: Q50968), Rickettsia conorii (ID: UVRA_RICCN AC: Q92G31),
Rickettsia prowazekii (ID: UVRA_RICPR AC: Q9ZCC3), Thermus thermophilus (ID: UVRA_THET8 AC: Q56242).
Следующая таблица является результатом сравнения двух выравниваний с помощью написанной мною программой (MSA.ipynb).
| Программа 1 | Программа 2 | Длина и процент выравненных колонок от общей длины для программы 1 | Длина и процент выравненных колонок от общей длины для программы 2 | Блоки | Индивидуально выравненные колонки |
|---|---|---|---|---|---|
| MAFFT | MUSCLE | 993, 94.46% | 990, 94.75% | 3,7=3,7 13,252=13,252 254,260=254,260 262,378=262,378 394,452=392,450 454,455=452,453 457,603=455,601 611,698=608,695 701,959=698,956 961,973=959,971 984,985=982,983 |
- | MAFFT | CLUSTALW | 993, 92.15% | 989, 92.52% | 1,316=1,316 319,333=319,333 340,375=340,375 394,452=392,450 454,455=452,453 457,477=455,475 484,603=482,601 611,696=608,693 701,958=698,955 984,985=981,982 |
- |
UvrA белки консервативны, потому по ним трудно судить о разнице выравниваний, но даже так, выравнивания схожи меньше, чем на 95%, и выравнивание, сделанное MAFFT, больше похоже на выравнивание, сделанное MUSCLE, чем CLUSTALW (2% cхожесть отличается). Возможно разница ClustalW от MAFFT связана с разительно отличающимися подходами к выравниваниям, у ClustalW оно прогрессивное, у MAFFT выравнивание сделано алгоритмом, основанном тоже на прогрессивном выравнивании, но при котором выравнивание проходит проверку с помощью рафинирования.
Ссылка на Jalview project с визуализированными выравниваниями: UVRA_MSA.jvp (в проекте 3 окна, с соответсвующими названиями в самой верхней строчке представленны 3 выравнивания).
Сравнение структурного выравнивания и множественного выравнивания, сделанного программой
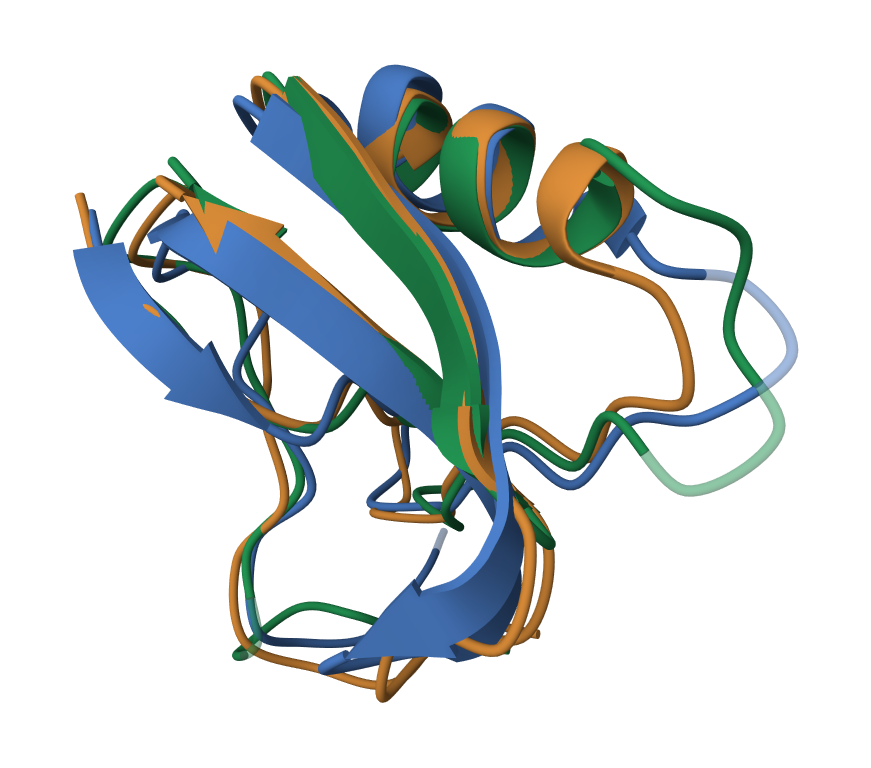
Для сравнения разных выравниваний возьмем 3 белка, содержащих Scorpion toxin-like domain (AC: PF00537): токсин скорпиона из Androctonus australis hector (PDB ID: 1AHO), токсин из Olivierus martensii (PDB ID: 2KBK), нейротоксин (TS1) из бразильского скорпиона Tityus Serrulatus (PDB ID: 1B7D).
Для сравнения выравниваний (структурное было сделанно следующим образом: 1aho по очереди было выравнено с 2kbk и 1b7d, затем вручную были добавлены недостающие гэпы) воспользуемся той же программой, что и в предыдущем блоке (см. табл. 1), ссылка на сами выравнивания: alignments.jvp (в проекте 2 выравнивания, в окне PDB - структурное, в другом - сделанное программой).
Выравнивания имеют одинаковую длину (см табл.2 ), но при этом сами выравнивания схожи лишь немногим больше, чем на 3 четверти (77.94%), сравнивания с результатами из прошлого пункта (см. табл 1, выравнивания, сделанные разными программами схожи на более, чем 90%).
| Программа 1 | Программа 2 | Длина и процент выравненных колонок от общей длины для программы 1 | Длина и процент выравненных колонок от общей длины для программы 2 | Блоки | Индивидуально выравненные колонки |
|---|---|---|---|---|---|
| MAFFT | Структурное выравнивание | 68, 77.94% | 68, 77.94% | 1,16=1,16 21,42=21,42 44,45=44,45 47,59=47,59 |
- |
Структуры белков (см рис. 1) довольно консервативны: бета-лист - альфа спираль - 2 бета-листа. Структурное выравнивание сохраняет это сходство и ставит гэпы в положениях 40-44 в 1B7D между двумя бета-листами, MAFFT нарушает это и затрагивает концы бета-листов, нарушая их.
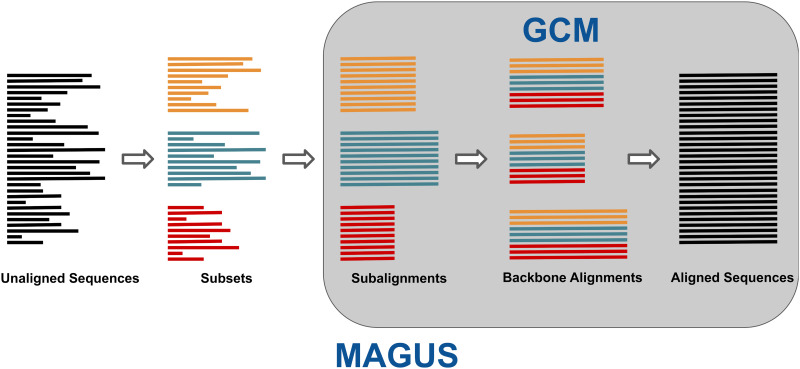
Описание MAGUS
Описание основы MAGUS
Многие алгоритмы множественного выравнивания не справляются с большими наборами последовательностей. MAGUS[1] - метод для решения этой проблемы.
Алгоритм MAGUS наследует базовую структуру более раннего алгоритма PASTA[2]. То есть MAGUS разбивает сиквенсы на подгруппы, выравнивает их (алгоритмом MAFFT-linsi[3]) , затем используя алгоритм объединения кластеров графов (GCM[4]) объединяет выравнивания (см. рис. 2).
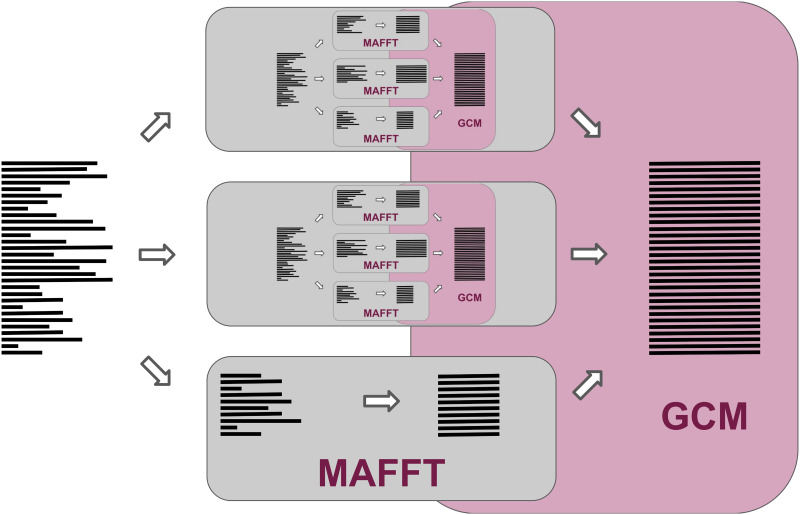
Улучшения MAGUS
Для увеличения показателей скорости и качества MAGUS использует некоторые улучшения. Одно из них это рекурсия: при разбивании сиквенсов на группы, если группа содержит сиквенсы больше порогового значения к ним применяется алгоритм MAGUS, если меньше - MAFFT (cм рис. 3).
Guide tree может быть построенно 4 способами:
1. 300 случайных последовательнсотей выравниваются с помощью MAFFT, а далее сиквенсы присоединяются с помощью эвристического алгоритма HMMER. Само дерево строится с помощью FastTREE
(такой же алгоритм в PASTA).
2. Метод построения начального дерева CLustalOmega.
3. Метод построения начального дерева MAFFT PartTree.
4. Метод построения минимального эволюционного дерева FastTree
Также MAGUS использует алгоритмы параллелизма и сжатия данных для увлеичения скорости.
Литература
- Smirnov V. Recursive MAGUS: Scalable and accurate multiple sequence alignment. PLoS Comput Biol. 2021 Oct 6;17(10):e1008950. doi: 10.1371/journal.pcbi.1008950. PMID: 34613974; PMCID: PMC8523058.
- Mirarab S, Nguyen N, Guo S, Wang LS, Kim J, Warnow T. PASTA: ultra-large multiple sequence alignment for nucleotide and amino-acid sequences. Journal of Computational Biology. 2015;22(5):377–386. doi: 10.1089/cmb.2014.0156
- Katoh K, Kuma K, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005 Jan 20;33(2):511-8. doi: 10.1093/nar/gki198. PMID: 15661851; PMCID: PMC548345.
- Smirnov V, Warnow T. MAGUS: Multiple sequence Alignment using Graph clUStering. Bioinformatics. 2021 Jul 19;37(12):1666-1672. doi: 10.1093/bioinformatics/btaa992. PMID: 33252662; PMCID: PMC8289385.
- Ссылка на MAGUS: MAGUS.