КОМПЛЕКС ДНК-БЕЛОК |
Назад |
define:
define set1 *.o?' and dna
define set2 *.op? and dna
define set3 *.N? and dna
 |
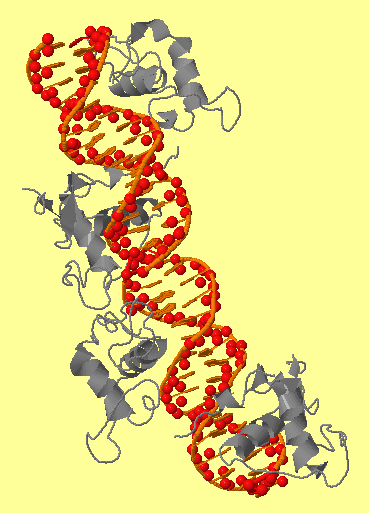 |
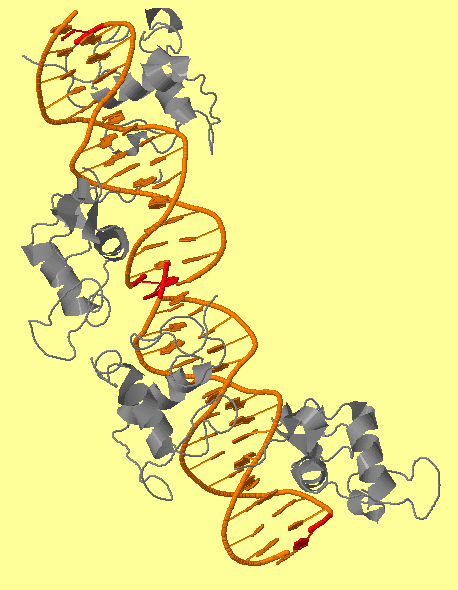
| фиг.1 — ДНК-белковый комплекс | фиг.2 — set1, кислороды при дезоксирибозе |
 |
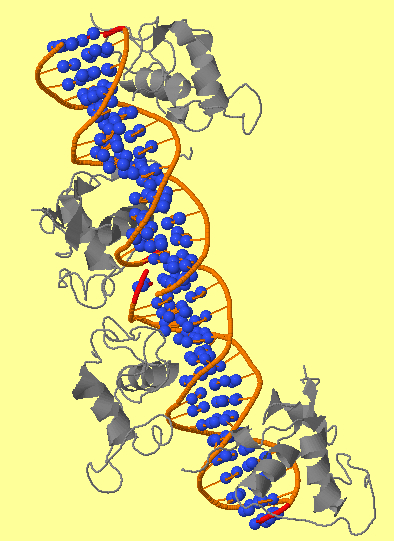 |
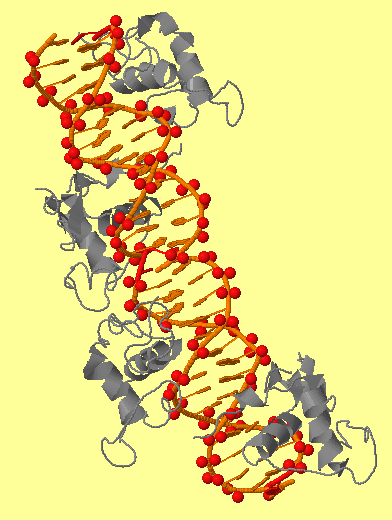
| фиг.3 — set2, кислороды при фосфатах | фиг.4 — set3, азоты при азотистых основаниях |
| Скрипт Jmol для последовательного отображения фиг.1-4: |
Скрипт Jmol для последовательного отображения того же, но в проволочной модели: |
background [xFFFF99]
|
background [xFFFF99]
|
Назовем полярным контактом ситуацию, в которой расстояние между полярным атомом белка и полярным атомом ДНК меньше 3.5A. Аналогично, неполярным контактом будем считать пару неполярных атомов на расстоянии меньше 4.5A.
Заполним табл.1:
Контакты атомов белка с |
Полярные |
Неполярные |
Всего |
остатками 2'-дезоксирибозы |
13 | 33 | 46 |
остатками фосфорной кислоты |
51 | 45 | 96 |
остатками азотистых оснований со стороны большой бороздки |
9 | 13 | 22 |
остатками азотистых оснований со стороны малой бороздки |
1 | 0 | 1 |
Всего |
74 | 91 | 165 |
Как можно видеть из табл.1, во взаиможействии между белком и ДНК наибольшую роль играют остатки фосфорной кислоты в остове ДНК, причём в то же время большая часть контактов неполярна.
nucplot получим схему взаимодействий белка и ДНК (фиг.5), предварительно переведя файл 1BY4.pdb в старый формат
командой remediator.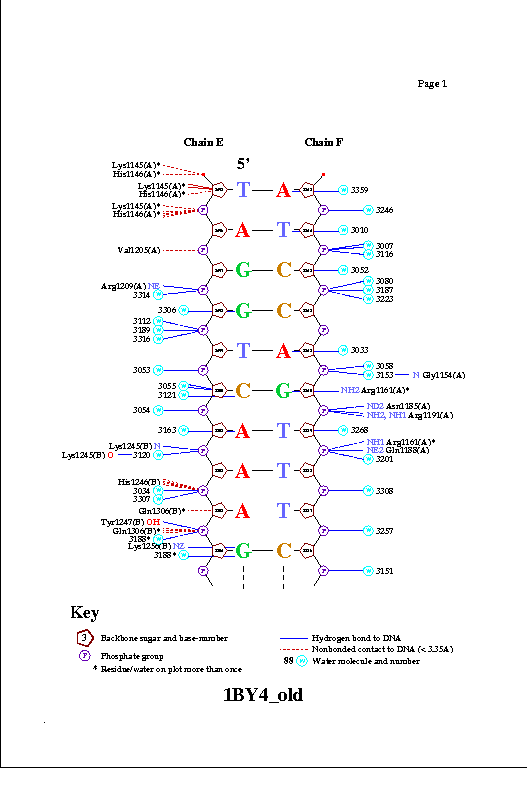
фиг.5 — схема контактов ДНК с водой и белком, страница 1 из 4. Полный перечень контактов
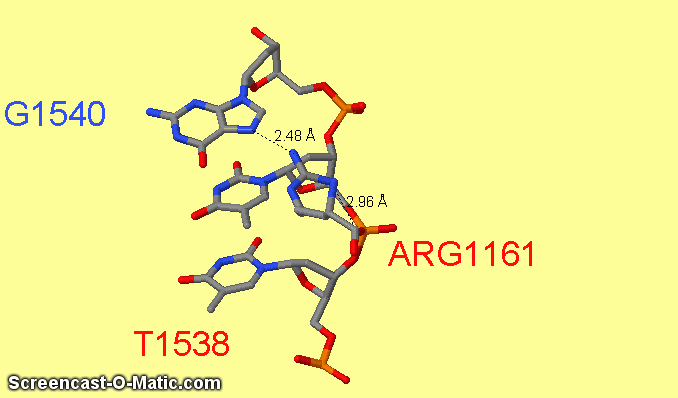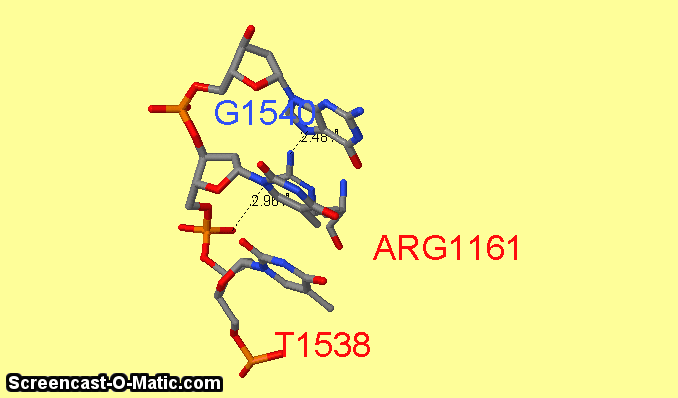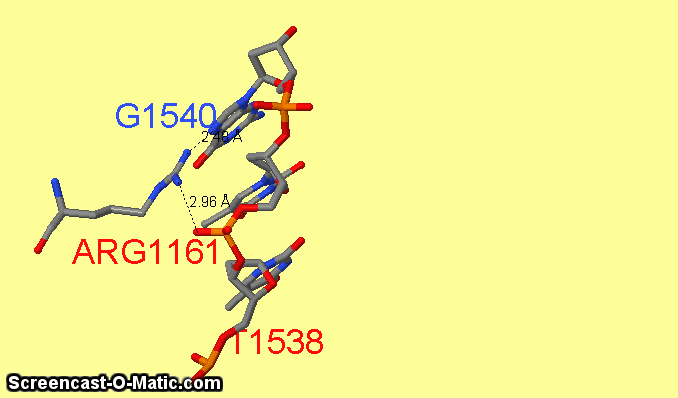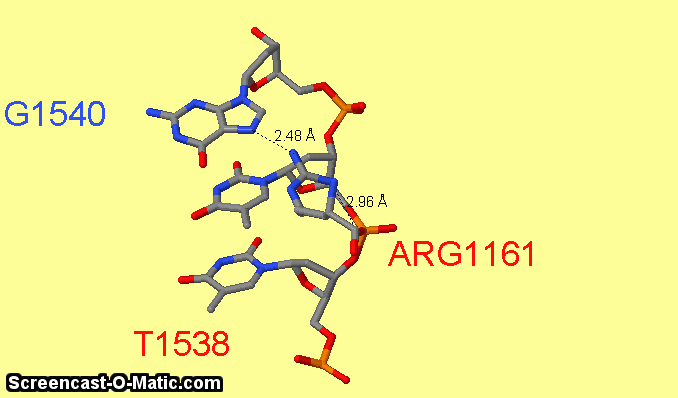
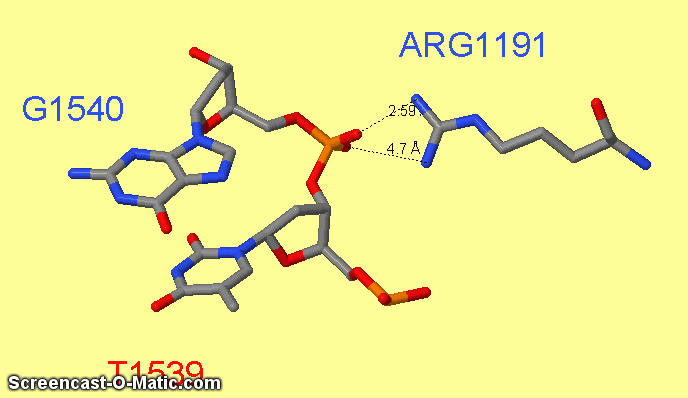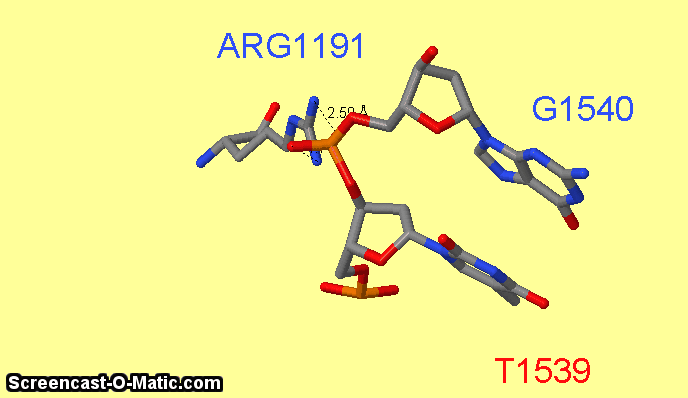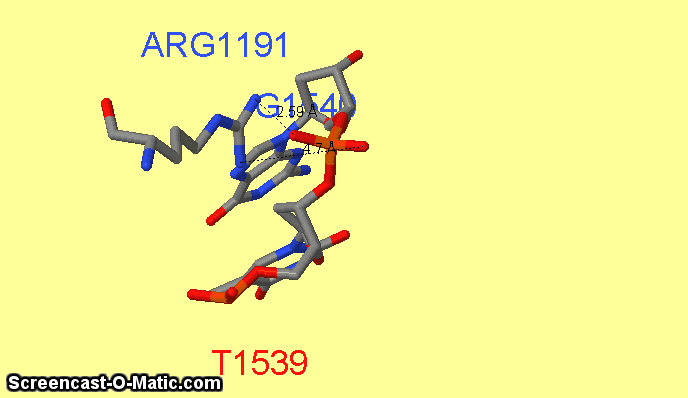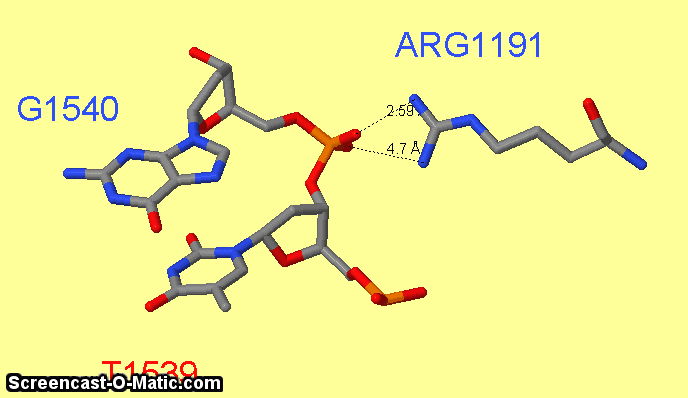
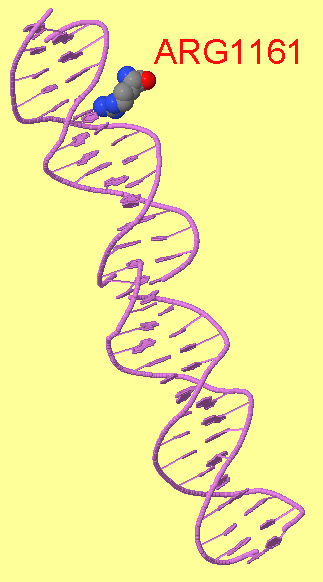
Как можно увидеть ARG1161 образует сразу два контакта с ДНК: с седьмым азотом при гуанине-1540 и кислородом фосфата при 3'-конце тимидина-1538 (см.фиг.6,8). Ещё одним важным для распознания ДНК аминокислотным остатком является ARG1191 (см.фиг.7,9), который неподалёку связывается с фосфатом.
{kind=link}
фиг.6 — контакты ARG1161 с ДНК; скрипт для получения этой анимации:
background [xFFFF99]
rotate x 90; rotate z 25; zoom out
zoomTo (atomindex=3065)
select arg1161
spacefill
color cpk
restrict arg1161 or dna
select atomno=225; label ARG1161; set fontsize 30
pause;
restrict 1540 or arg1161 or 1538 or 1539
measure 3062 231
measure 3093 232
select atomno=3099; label G1540
set fontsize 30
set labeloffset 15 0
select atomno=3056; label T1538; set fontsize 30; set labeloffset 0 -36
move 0 360 0 0 0 0 0 0 8
{kind=link}
фиг.7 — контакты ARG1191 с ДНК; скрипт для получения этой анимации:
background [xFFFF99]
select arg1191 or dna; cartoons off; wireframe 50; color cpk
measure 490 3076
measure 489 3075
restrict arg1191 or 1540 or 1539
select atomno=3099; label G1540; set fontsize 30
set labeloffset 0 40
select atomno=3076; label T1539; set fontsize 30;
set labeloffset 0 -40
select atomno=488; label ARG1191; set fontsize 30; set labeloffset 0 30
select atomno=488; label ARG1191; set fontsize 30; set labeloffset 0 50
zoomTo (atomindex=3074)
move 0 360 0 0 0 0 0 0 8
 |
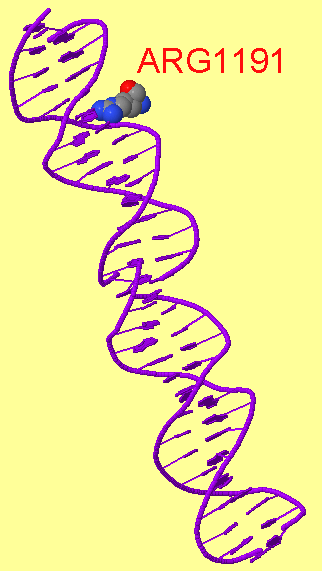 |
| фиг.8 — положение ARG1161 относительно ДНК | фиг.9 — положение ARG1191 относительно ДНК |
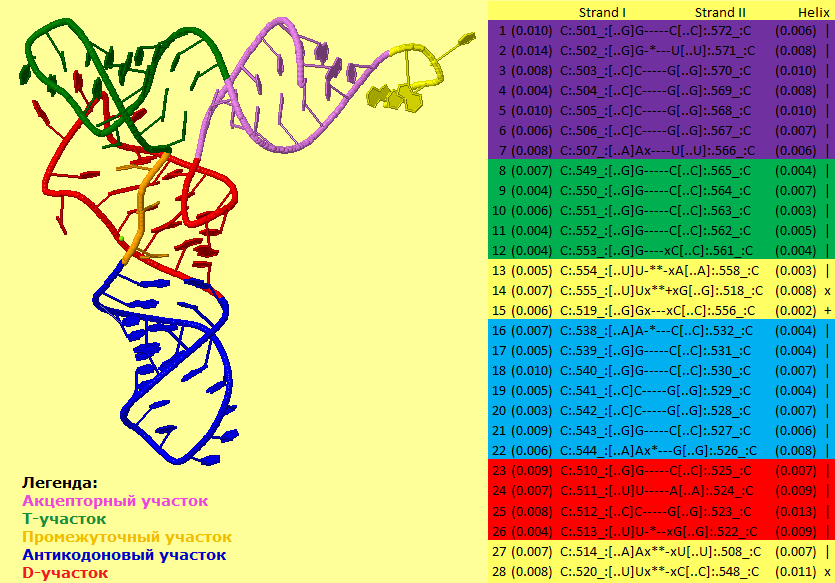
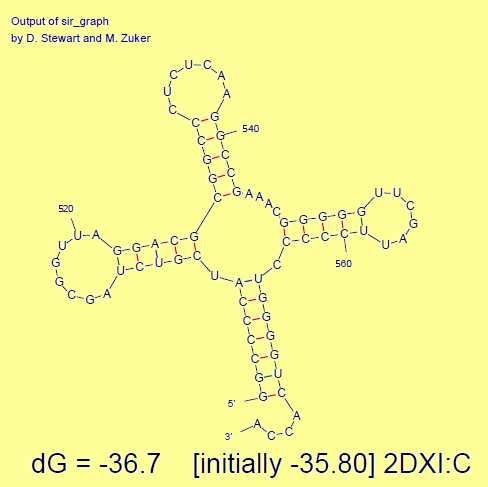
фиг.9 — структура тРНК, справа таблица, полученная командой
find_pair с выделенными стеблями.Cкрипт для получения изображения в Jmol:
background [xFFFF99]
restrict :c; cpk off; wireframe off; cartoons on
select 573-576; color yellow
select 501-507 or 566-572; color violet
select; cartoons off; wireframe 35
select 549-565; color green
select 526-544; color blue
select 508-525; color red
select 545-548
color orange
Применим поиск выравниваний с помощью команды einverted. Параметры поиска:Gap penalty [12]: 12
Minimum score threshold [50]: 15 (при более высоких значениях выравнивания не находятся)
Match score [3]: 3
Mismatch score [-4]: -3 (при стандартном значении находится только одно выравнивание)
Вывод программы имеет следующий вид:
SEQUENCE: Score 15: 7/9 ( 77%) matches, 0 gaps
1 ggccccatc 9
|||| | ||
31 ccggcgcag 23
SEQUENCE: Score 15: 10/15 ( 66%) matches, 0 gaps
34 ctcaaggccgaaacg 48
| | ||| ||| ||
69 ggggtcccccttagc 55
Если всё же выставить Mismatch score [-4], то вывод будет выглядеть так:
SEQUENCE: Score 15: 5/5 (100%) matches, 0 gaps
3 cccca 7
|||||
69 ggggt 65
То есть удаётся найти только пять из семи пар акцепторного стебля.
| Участок структуры |
Позиции в структуре (по результатам find_pair) |
Успешно предсказанные пары с помощью einverted |
Успешно предсказанные пары по алгоритму Цукера |
| Акцепторный стебель | 7 пар 5'501-507 3' 3' 572-566 5' |
5 | 7 |
| D-стебель | 4 пары 5' 510-513 3' 3' 525-522 5' |
0 | 4 (+1 ошибочно) |
| T-стебель | 5 пар 5' 549-553 3' 3' 565-561 5' |
0 | 4 (+1 ошибочно) |
| Антикодоновый стебель | 7 пар 5' 526-532 3' 3' 544-538 5' |
0 | 5 (1 из пропущенных пар включена в D-стебель) |
| Общее число угаданных пар нуклеотидов | 23 | 5 | 20 |
табл.2 — сравнение автоматизированных методов определения структуры тРНК
Как легко заметить, алгоритм Цукера является очень точным способом определения структуры тРНК (см.фиг.10) по последовательности нуклеотидов. При поиске структуры допустимым отклонение по энергии Гиббса от минимальной было установлено в 5%. Использовалась онлайн-версия алгоритма.