| Главная | Семестры | Проекты | Заметки | О себе | Полезные ссылки |
Выбор домена
Условия выбора домена: не более 10000 последовательностей из не более 1200 видов, минимум 2 разные доменные архитектуры (каждая представена в >20 последовательностях), хотя бы для одной последовательности известна 3D структура белка (домена). По всем этим критериям проходит Formin Homology 2 Domain (Formins).Описание домена:
| AC | PF02181 |
| Функция домена: | Cемейство форминов вовлечено в полимеризацию актина и связан с активно растущим концом актинового филамента. С помощью FH2 домена формины напрямую связываются с микротрубочками. |
| Страница домена в Pfam | Ссылка |
| Число видов | 317 |
| Число последовательностей: | 2361 |
| Число архитектур: | 102 |
| Список архитектур, в которые входит домен кинезин с числом последовательностей | Ссылка |
Затем получим выравнивания последовательностей, содержащих наш домен. Для этого воспользуемся программой JalView. Последовательность действий: скачаем из Pfam с помощью JalView наши последовательности (File - Fetch). Затем раскрасим их по консервативности (ClustalX и By conservation с порогом консервативности 10%). Добавим 3D структуру домена (Uniprot: BNI1_YEAST и DAAM1_HUMAN, PDB: соответственно 1UX4 и 2Z6E). Полученные проекты выравнивания в форматах:
FASTA и JALVIEW
Далее получим сводную таблицу, содержащую информацию о последовательностях всех белков, содержащих наш домен. Для этого воспользуемся соответствующими скриптами: sw_to_xls.py и uniprot_to_taxonomy.py.
В putty введем следующие команды:
python sw_to_xls.py -p PF02181 -i /srv/databases/pfam/swisspfam.gz -z -o swisspfam_to_xls_out.txt
python uniprot_to_tax.py -i tax.txt -o out.txt
Ссылка на сводную таблицу Excel с описанием всех белков из Uniprot, включающих домен (лист - "сводная таблица", включает в себя ID последовательностей, AC Pfam доменов, которые к ним относятся, а также таксономию последовательностей и длину домена FH2).
Выбор архитектур
Таблица 1. Выбранные архитектуры Pfam| Изображение | Архитектура | Число представителей | Описание других доменов |
| Drf_FH3, Drf_GBD, FH2 | 415 | Drf_FH3 - встречается в формин-подобных и DIAPH белках, Drf_GBD - домен, который взаимодействует с ГТФ-связаным Rho-белком, что приводит к активации белка DRF (из семейства Diaphanous related formins). | |
 |
FH2 | 1001 | - |
Выбор таксона и подтаксонов
Для изучения указанных выше архитектур был выбран таксон Metazoa с подтаксонами Ecdysozoa и Chordata. Metazoa (другие названия Animalia, Animals) - царство многоклеточных, эукариотических организмов, основными признаками которых являются гетеротрофность и способность к акивному передвижению. Подтаксон Chordata - тип вторичноротых животных, для которых характерно наличие энтодермального осевого скелета в виде хорды, которая у высших форм заменяется позвоночником. Ecdysozoa - группа первичноротых животных. Предложен на основе молекулярно-генетических исследований генов рРНК малых субъединиц рибосом [1].Работа с выборкой и выравниванием
Выборка представлена в ТАБЛИЦЕ на листе "выборка". Также были добавлены 2 последовательности с известной 3D структурой: BNI1_YEAST и DAAM1_HUMAN. Отфильтруем последовательности с помощью скрипта filter_alignment.py, используя команду:python filter_align.py -i file.fasta -m ids.txt -o filter_alignment_out.fasta
Обозначим наши таксоны сокращенно: (C)-Chordata, (E)-Ecdysozoa.
Обозначим архитектуры: 1 - доменная архитектура FH2, 2 - доменная архитектура Drf_FH3, Drf_GBD, FH2. В названия последовательностей добавим наши сокращения, например, если последовательность относится к первой архитектуре (FH2) и таксону Chordata, то перед ее названием будет стоять С1. Соответственно, если последовательность будет относиться ко второй архитектуре (Drf_FH3, Drf_GBD, FH2) и такону Ecdysozoa, то перед названием будет стоять E2.
Полученный файл откроем в Jalview. Удалим пустые колонки, создадим две группы (группа 1 - FH2, группа 2 - Drf_FH3, Drf_GBD, FH2). Зададим раскраску в каждой группе (ClustalX+By conservation, порог = 5%). Добавим разметку, исходя из вторичной структуры наших последовательностей BNI1_YEAST и DAAM1_HUMAN. Удалим явно плохо выровненные последовательности: E2R031_CANFA, Q8MT16_DROME, H3AGL2_LATCH, D3ZP34_RAT, G3T1K4_LOXAF, G6D451_DANPL, A2T750_PANTR, BNI1_YEAST. Остается 33 последовательности. Разметим вторичную структуру, используя последовательность DAAM1_HUMAN (альфа-спирали отмечены в Secondary Structure красным цветом). Удалим N- и C-концевые части выравнивания, не несущие смысла. Представленное выравнивание сохраняем в формате .jar: project_2.jar.
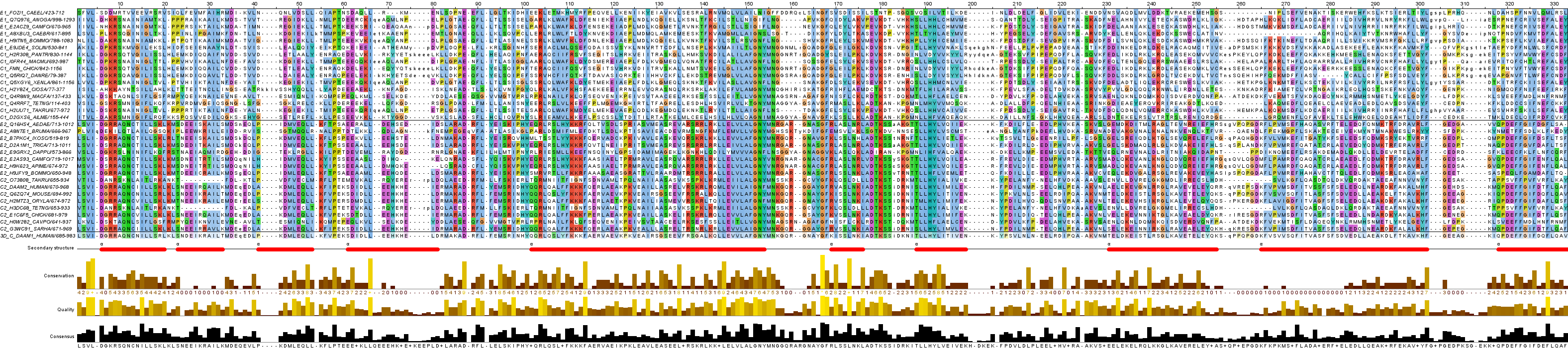
Рис. 1. Выравнивание домена выбранных последовательностей (изображение кликабельно).
Построение дерева с помощью выравнивания
Дерево получим с помощью программы MEGA методом Neighbor-joining (метод, который не использует гипотезу о молекулярных часах и строит неукоренённое дерево). Применим bootstrap-анализ при построении дерева, число реплик - 100: tree_bootstrap.nwk.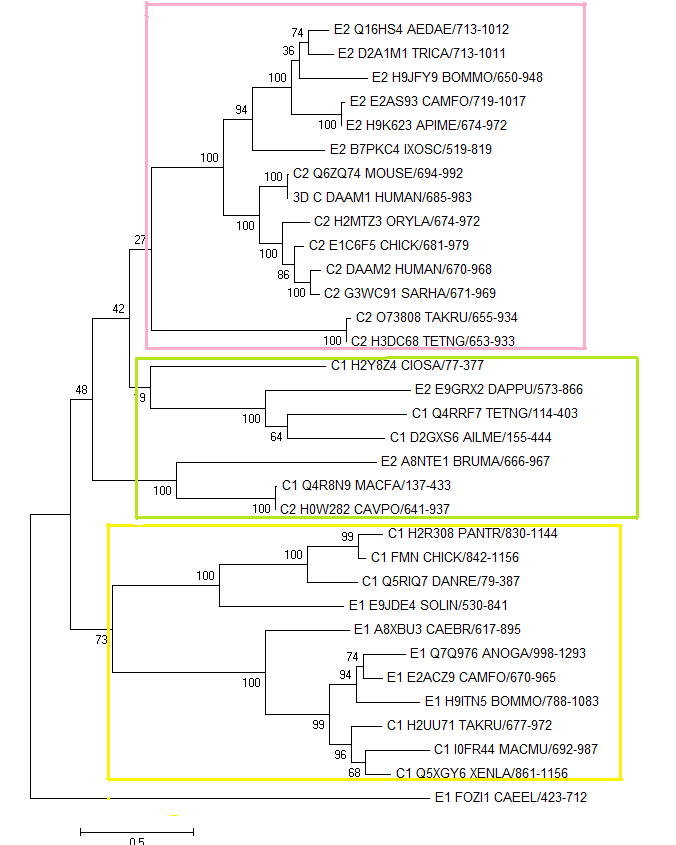
Рис. 2. Изображение дерева, полученного с помощью bootstrap-анализа. Розовым выделена клада, относящаяся ко второй доменной архитектуре, желтым - к первой. Зеленым выделены ветви, в которые попали последовательности и из первой, и из второй архитектуры.
1)В целом, прослеживаются две клады, отвечающие за первую и вторую архитектуру, однако есть выбивающаяся совсем ветвь FOZI1_CAEEL (Caenorhabditis elegans, Ecdysozoa), а также проблемный участок, обведенный зеленым на рисунке 2. Мы можем предположить, что разделение доменных архитектур в эволюции произошло давно, т.е. общий предок Ecdysozoa и Chordata имел обе архитектуры. Попробуем выяснить, почему у нас возникла проблемная ветвь.
В эту ветвь попали последовательности:
E2_E9GRX2_DAPPU/474-867 (Daphnia pulex (Water flea))
C2_H0W282_CAVPO/563-938 (Cavia porcellus (Guinea pig))
E2_A8NTE1_BRUMA/586-968 (Brugia malayi (Filarial nematode worm))(архитектура 2)
C1_H2Y8Z4_CIOSA/6-378 (Ciona savignyi (Pacific transparent sea squirt))
C1_Q4RRF7_TETNG/36-404 (Tetraodon nigroviridis (Spotted green pufferfish))
C1_D2GXS6_AILME/75-445 (Ailuropoda melanoleuca (Giant panda))
C1_Q4R8N9_MACFA/57-434 (Macaca fascicularis (Crab-eating macaque) (Cynomolgus monkey))(архитектура 1).
Эта "проблемная ветвь" должна по идее относится ко второй архитетуре. В этой ветви четко прослеживается таксономическое деление (как будто бы сначала произошло разделение на таксоны, а уже в каждом из них архитектуры появились независимо). Причем эти ветви имеют хорошую поддержку. Быть может, имеет смысл говорить об ошибке в выборке, либо о повторном образовании архитектур.
2)Также можно отметить, что в подтаксонах последовательности хорошо разделяются по принадлежности к архитектурам.
3)Анализ bootsrap показал, что ветви, отделяющие последовательности с двумя разными архитектурами, имеют относительно слабую поддержку. Но очень хорошую поддержку (100 деревьев содержали эти ветви) имеют те ветви, которые отделяют последовательности, принадлежащие одному таксону.
Попробуем убрать "выбивающиеся" последовательности из первой архитектуры в проблемой ветви. Построим дерево с помощью алгоритма максимального правдоподобия. Полученное дерево все равно имеет те же ветви, содержащие представителей разных архитектур. Быть может, это действительно связано со вторичным образованием (дупликацией гена).

Рис. 3. Дерево, построенное с помощью алгоритма максимального правдоподобия.
Здесь можно также заметить, что одна группа организмов выбрала сложный домен (архитектура 2, усложнение простого домена), а другая - простую однодоменную архитектуру 1. Однако, есть организмы, которые предпочли первую простую архитектуру (ветвь с поддержкой 22). Возможно в них она возникла независимо от сложной архитектуры. Здесь бутстреп-анализ также показал очень высокие значения поддержки ветвей.