Программмы выравнивания.
Сравнение трех программ выравнивания:
Tcoffee, Muscle, Mafft
Для сравнения выравниваний я решил выбрать последовательности субъединицы D1
фотососистемы II из 5 далеких, в таксономическом плане, организмов: Arabidopsis thaliana,
Chara vulgaris, Chlamydomonas reinhardtii, Acetabularia acetabulum,
Euglena gracilis, Nostoc sp. . Была выбрана субъединица фотосистемы, так как, по предположению,
в связи с ее важной ролью в фотосинтезе вышеперечисленных организмов, она должна быть консервативной
Проект JalView
Оказалось, что данный белок чересчур консервативен: даже в таких дальних организмах
последовательность практически идентична. Однако, даже в данном случае удалось сделать
интересное наблюдение: при помощи программы acab Ивана Петрушко были сравнены
3 выравнивания, выполненные Tcoffee, Mafft, Muscle. Оказалось, что выравнивания
Mafft и Muscle получились идентичными, а у Tcoffee совпадают колонки 4-359 (98.61%).
Чтобы сравнить программы лучше, было решено выбрать менее консервативные
белки: в итоге была выбрана фруктозо-1,6-бисфосфатальдолаза (EC 4.1.2.13) - фермент участвующий
в процессе гликолиза.
Проект JalView
Были выравнены 4 последовательности из следующих организмов:
Homo sapiens, Escherihia coli, Nostoc commune, Arabidopsis thaliana. Результаты
приведены в таблицах 1, 2 и 3.
| Начало в Tcoffee | Конец в Tcoffee | Начало в Muscle | Конец в Muscle |
|---|---|---|---|
| 49 | 61 | 43 | 55 |
| 96 | 97 | 90 | 91 |
| 213 | 236 | 167 | 190 |
| 270 | 270 | 230 | 230 |
| 371 | 377 | 282 | 288 |
| 382 | 391 | 293 | 302 |
| 425 | 425 | 327 | 327 |
| 514 | 515 | 415 | 416 |
| Начало в Tcoffee | Конец в Tcoffee | Начало в Mafft | Конец в Mafft |
|---|---|---|---|
| 65 | 67 | 47 | 49 |
| 75 | 88 | 57 | 70 |
| 97 | 101 | 79 | 83 |
| 128 | 129 | 133 | 134 |
| 132 | 157 | 137 | 162 |
| 197 | 201 | 200 | 204 |
| 215 | 243 | 218 | 246 |
| 271 | 271 | 282 | 282 |
| 282 | 284 | 293 | 295 |
| 294 | 294 | 257 | 257 |
| 352 | 357 | 336 | 341 |
| 366 | 368 | 351 | 353 |
| 378 | 381 | 365 | 368 |
| 411 | 413 | 414 | 416 |
| 462 | 462 | 461 | 461 |
| 514 | 515 | 507 | 508 |
| Начало в Muscle | Конец в Muscle | Начало в Mafft | Конец в Mafft |
|---|---|---|---|
| 91 | 91 | 79 | 79 |
| 169 | 190 | 218 | 239 |
| 213 | 213 | 264 | 264 |
| 304 | 315 | 380 | 391 |
| 322 | 324 | 398 | 400 |
| 338 | 345 | 417 | 424 |
| 357 | 357 | 436 | 436 |
| 401 | 417 | 493 | 509 |
При сравнении выравниваний субъединицы D1 фотосистемы II, как уже писалось ранее, оказалось, что Mafft и Muscle выдали одинаковые выравнивания, в отличие от Tcofee. Из этого можно было сделать вывод, что их алгоритмы более схожи, чем алгоритм Tcofee. Однако уже в примере с альдолазой результат другой: Tcofee и Mafft имеют наибольший процент общих колонок (20.93% от выравнивания Tcoffee; 21.22% от выравнивания Mafft). В это же время при остальных сравнениях процент общих колонок не превышает 15.60%. Рассмотрим отдельный пример блока, имеющегося, во всех выравниваниях: это, например. 218 - 239 колонки в выравнивании Mafft. Так как он присутствует в выравнивании каждой из программ, а цель каждой из программ - построить выравнивание, близкое к эволюционному, то про колонки, входящие в данный блок, можно с большей уверенностью говорить, что они выровнены эволюционно.
Выравнивание по совмещению структур
Для выравнивания по совмещению структур я выбрал 3 белка семейства IS200 похожих
транспозаз(PF01797), имеющих структуру в базе данных PDB:
- Q933Z0_HELPX (PDB: 2a6m)
- Q974H8_SULTO (PDB: 2ec2)
- Q97Y68_SACS2 (PDB: 2f4f)
Я получил 2 выравнивания трех этих белков: одно с помощью выравнивания по 3D-структуре при помощи сервиса Pairwise Structure Alignment на сайте PDB, и второе - при помощи программы Muscle. На рисунке 1 представлено изображение совмещения трех структур.
Проект JalView
При помощи программы acab Ивана Петрушко, я сравнил эти два выравнивания. Результаты приведены в таблице 4.
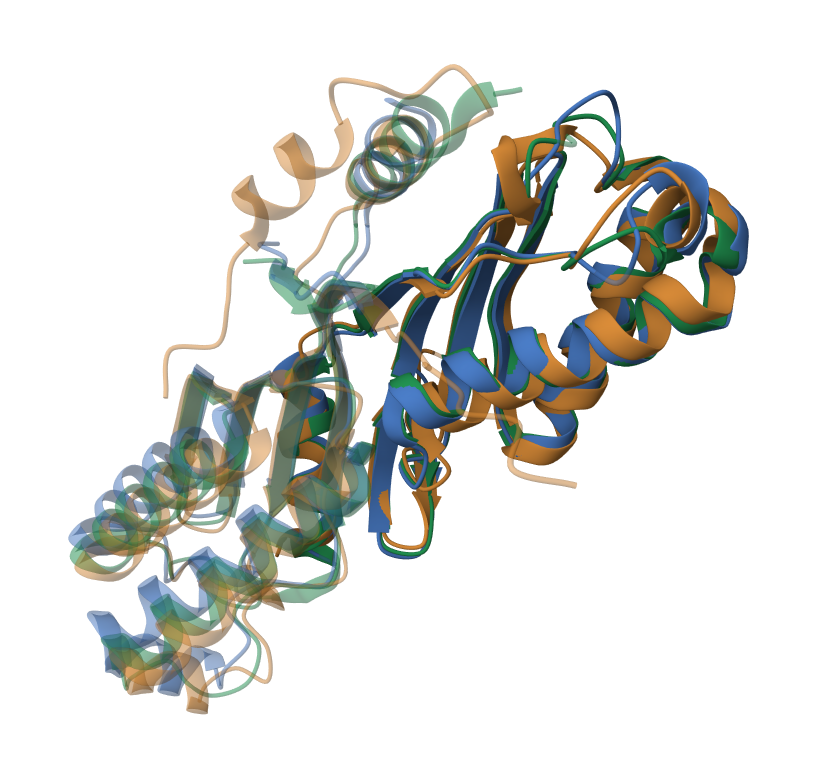
| Начало в Muscle | Конец в Muscle | Начало в 3D-совмещении | Конец в 3D-совмещении |
|---|---|---|---|
| 1 | 97 | 1 | 97 |
| 99 | 99 | 99 | 99 |
| 103 | 135 | 103 | 135 |
Процент совпадающих колонок в этих выравниваниях - 97.04%. Как несложно заметить из таблицы, действительно, выравнивания практически идентичны.
Программа множественного выравнивания последовательностей Muscle.
MUSCLE (Multiple Sequence Comparison by Log-Expectation) — это алгоритм для множественного выравнивания белковых последовательностей. [1]
Прежде чем говорить о работе алгоритма, стоит сказать о двух мерах сравнения
расстояния при построении направляющих деревьев [1]:
- Расстояние K-меров (Kmer distance). K-мер - непрерывная подпоследовательность длины K. Расстояние K-меров определяется долей общих K-меров двух последовательностей. Расстояние K-меров позволяет дать оценку сходства последовательностей без их выравнивания, что позволяет экономить время.
- Расстояние Кимури (Kimura distance) - оценка сходства выравненных последовательной с поправкой на возможное множественных замен в процессе эволюции одной аминокислоты.
- Этап 1. При помощи расстояния K-меров строится бинарное дерево TREE1, где листья дерева - последовательности, а каждый узел - есть выравнивание. То есть корень дерева TREE1 является множественным выравниванием MSA1 всех Введенных последовательностей.
- Этап 2. Источником неоптимального выравнивания на предыдущем этапе является приближенная оценка расстояний K-меров. Но после первого этапа у нас есть выравнивание: теперь мы можем воспользоваться расстоянием Кимури. Для каждой пары последовательностей из MSA1 вычисляется расстояние Кимури, на основе которого строится бинарное дерево TREE2. Как и на этапе 1, корень этого дерева является множественным выравниванием MSA2 всех последовательностей.
- Этап 3. Выбирается ребро (начинаем с ближайшего к корню дерева). TREE2 делится на 2 поддерева путем удаления этого ребра. Новое множественное Выравнивание получается путем выравнивания профилей находящихся в корнях этих двух деревьев. Если выравнивание получилось лучше - то оно сохраняется. Этот процесс повторяется сначала до тех пор, пока выравнивание не перестает улучшаться или достигается заданный на входе предел. Таким образом мы получаем окончательное множественное выравнивание MSA3.
Именно за счет наличия нескольких этапов алгоритм Muscle дает хороший баланс между скоростью выполнения и эффективностью нахождения оптимального множественного выравнивания.
СПИСОК ЛИТЕРАТУРЫ
[1] Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004 Mar 19;32(5):1792-7.