Сборка de nova генома Buchnera aphidicola
Подготовка и очистка чтений
Для сборки был использован набор single-end чтений длиной 39 нуклеотидов, полученных по технологии Illumina, секвенирования Buchnera aphidicola strain Tuc7 (ENA). Исходный файл полученных чтений был формате fastq.gz.
Для подготовки чтений нужно было провести их очистку: убрать некачественно секвенированные нуклеотиды
и оставшиеся адаптеры. Для адаптеров был создан объединенный файл с последовательностями
адаптеров из директории /mnt/scratch/NGS/adapters.
Очистка чтений проводилась программой TrimmomaticSE (то есть для одноконцевых чтений).
Сначала был запущен без дополнительных, чтобы оценить качество чтений.
TrimmomaticSE -threads 4 -phred33 SRR4240361.fastq.gz SRR4240361_trimmed.fastq.gz ILLUMINACLIP:adapters/adapters.fasta:2:7:7
Вывод Trimmomatic без параметров
Input Reads: 7272621
Surviving: 7238089 (99.53%)
Dropped: 34532 (0.47%)
На основании большого процента "выживших" чтений можно говорить о высоком качестве сборке и малом количестве остатков адаптеров. Но для сборки нужно отсеить более тщательно. Для этого TrimmomaticSE был запущен с параметрами: только чтения длиной не менее 32 нуклеотида и с качеством нуклеотидов справа не ниже 20.
TrimmomaticSE -threads 4 -phred33 SRR4240361.fastq.gz SRR4240361_trimmed_param.fastq.gz ILLUMINACLIP:adapters/adapters.fasta:2:7:7 TRAILING:20 MINLEN:32
Вывод Trimmomatic с параметрами
Input Reads: 7272621
Surviving: 6834335 (93.97%)
Dropped: 438286 (6.03%)
Чтений все еще осталось много, чего мы и ожидали от запуска без параметров.
Сборка генома программой Velveth
Для сборки генома de novo использовалась программа velvet.
Подготовка данных осуществлялась программой velveth с длиной k-мера (hash_length) 31.
Чтения рассматривались как короткие и непарные (short). Подготовка k-меров velveth
была выполнена при помощи следующей команды:
velveth velveth_31 31 -short -fastq.gz SRR4240361_trimmed_param.fastq.gz
Параметры velveth
-short - короткие непарные чтения
velveth_31 — директория для результата
31 — hash_length (длина k-меров)
-fastq.gz — формат входных данных
Сборка контигов из k-меров, подготовленных программой velvet выполнялась программой velvetg. Cборка была осуществленна при помощи следующей команды:
velvetg velveth_31 -exp_cov auto -cov_cutoff auto
Параметры velvetg
-exp_cov auto - автоматический подсчет ожидаемого покрытия
-cov_cutoff auto - автоматически отбрасывает контиги с низким покрытием
Анализ полученных контигов
Для сравнения был выбран референсный штамм Buchnera aphidicola str. Sg с записью
хромосомы в nuccore c id NC_004061.1.
Для анализа полученных контигов был написан python-скрипт.
Код скрипта python.
#!/usr/bin/env python3
import sys
import statistics
contigs_fa = sys.argv[1]
lengths = []
coverages = []
contigs = []
with open(contigs_fa) as f:
for line in f:
if line.startswith(">"):
header = line.strip()
parts = header.split("_")
length = int(parts[3])
coverage = float(parts[5])
lengths.append(length)
coverages.append(coverage)
contigs.append((length, coverage, header))
median_coverage = statistics.median(coverages)
# три самых длинных контига
top3 = sorted(contigs, reverse=True)[:3]
# N50
total_length = sum(lengths)
half = total_length / 2
lengths_sorted = sorted(lengths, reverse=True)
current_sum = 0
N50 = 0
for l in lengths_sorted:
current_sum += l
if current_sum >= half:
N50 = l
break
# аномально покрытые
low_cov = median_coverage / 5
high_cov = median_coverage * 5
anomal = [c for c in contigs if c[1] < low_cov or c[1] > high_cov]
# вывод
print("Общее число контигов:", len(contigs))
print("Суммарная длина сборки:", total_length)
print("N50:", N50)
print()
print("Три самых длинных контига:")
for l, c, h in top3:
print("\tдлина =", l, "покрытие =", round(c, 2), "контиг - ", h)
print()
print("Медианное покрытие:", rounad(median_coverage, 2))
print("Аномальное покрытие: <", round(low_cov, 2), "или >", round(high_cov, 2))
print("Контигм с аномальным покрытием:")
for i in range(len(anomal)):
print(" ", anomal[i][2])
Вывод скрипта python.
Общее число контигов:
81
Суммарная длина сборки:
652407
N50:30106
Три самых длинных контига:
длина = 49238 покрытие = 26.66 контиг - >NODE_6_length_49238_cov_26.660851
длина = 45555 покрытие = 26.45 контиг - >NODE_2_length_45555_cov_26.450466
длина = 43866 покрытие = 23.51 контиг - >NODE_28_length_43866_cov_23.514977
Медианное покрытие:
25.76
Аномальное покрытие: меньше 5.15 или больше 128.81
Контигм с аномальным покрытием:
>NODE_82_length_31_cov_216.354843
Как видно из вывода скрипта, N50 равен 30106, и лишь один контиг аномально покрыт (NODE_82).
Далее мы рассматриваем три контига с наибольшим покрытием: строим карты локального сходства с хромосомой Buchnera aphidicola другого штамма - Buchnera aphidicola str. Sg (Schizaphis graminum).
Контиг NODE_6
Контиг NODE_6 имеет длину 49238 п.н. и среднее покрытие ~26.7.
Карта локального сходства приведена на рисунке ниже. Локальные выравниваний соответствует участку хромосомы в координатах 130911–176511.
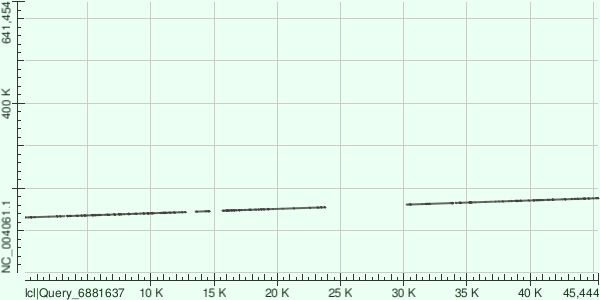
Hit Table: Hit Table для NODE_6
Контиг NODE_2
Контиг NODE_2 имеет длину 45555 п.н. и среднее покрытие ~26.5. Выровнялся с минус цепью данной хромосомы.
Локальные выравнивания соответствует участку хромосомы в диапазоне координат 449737–496030.

Hit Table:Hit Table для NODE_2
Контиг NODE_28
Контиг NODE_28 имеет длину 43866 п.н. и среднее покрытие ~23.5. Для него было обнаружено несколько коротких локальных сегментов сходства с референсным геномом.
Локальные выравнивания соответствует участку хромосомы с координатами 256307–298229.
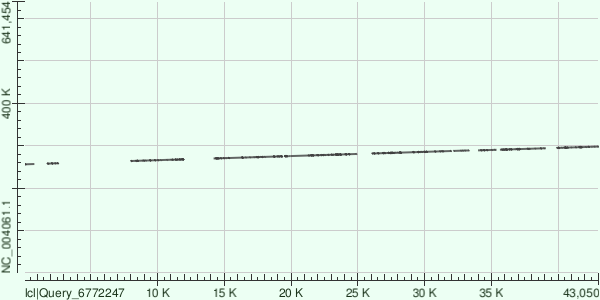
Hit Table: Hit Table для NODE_28
Выводы
Смотря на dotplot'ы с выравниями может прийти мысль о том, что пустые части - делеции или вставки.
Но это не так - такие участки длинной несколько тысяч нуклеотидов лишь показывают, что участки плохо выравнялись,
то есть наши участки не очень похожи (к тому же использовался чувствительный алгоритм megablast; более того,
данные участки и не обязаны быть консервативными).
Однако, стоит заметить, делеции и инсерции тоже могут присутствовать.
Еще можно заметить, что длина выровненного участка хромосомы меньше, чем длина контига - это можно связать с
малой схожестью на концах контига.