DNA and protein complexes
Task 1. Predicting the secondary structure of a given tRNA
The exercise was performed using the program einverted from the EMBOSS package.
the obtained complementary sites were compared with their descriptions obtained in the find_pair program
The following parameters were given as input to the program:
! einverted -sequence rna.seq -gap 5 -threshold 1 -match 2 -mismatch -3 -outfile tRNA3.inv -outseq seqout
The data obtained do not correspond to the real RNA structure. It was not possible to select parameters that reflect the real structure. This is probably due to the fact that the algorithms designed for DNA are not suitable for working with RNA.
Next, the secondary structure of tRNA was predicted using the Zucker algorithm (with ViennaRNA).
the resulting structure is shaped like a coverleaf
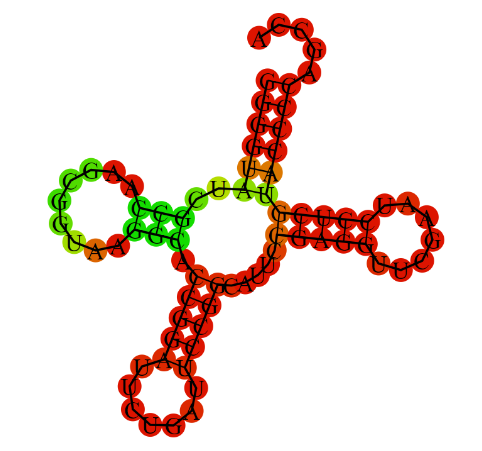
The results of the comparison of the find_pair and einverted programs are shown in the table below
| Structure section | Coordinates in the structure(find_pair results) | Prediction with einverted | Results of Zucker's algorithm prediction |
| Acceptor stem | 5' 2-7 3' 5' 66-71 3' |
6 pairs predicted | 6 pairs predicted |
| D-stable | 5' 10-12 3' 5' 23-25 3' |
- | 3 pairs predicted |
| T-stable | 5' 49-53 3' 5' 61-65 3' |
- | 5 pairs predicted |
| Anticodone stem | 5' 37-44 3' 5' 26-33 3' |
- | 5 pairs predicted |
| Total number of canonical nucleotide pairs | 19 | 19 | 6 |
Task 3
Coordinates of tRNA stems:
| Strand 1 | Strand 2 | |
| 2-7 | 66-71 | |
| 49-53 | 61-65 | |
| 37-44 | 26-33 | |
| 10-12 | 23-25 |
Non-canonical base pairs:
55U-18G
38U-32U
44C-26A
13A-45A
14A-21A
19G-56C is additional hydrogen bond that maintains the tertiary structure of tRNA
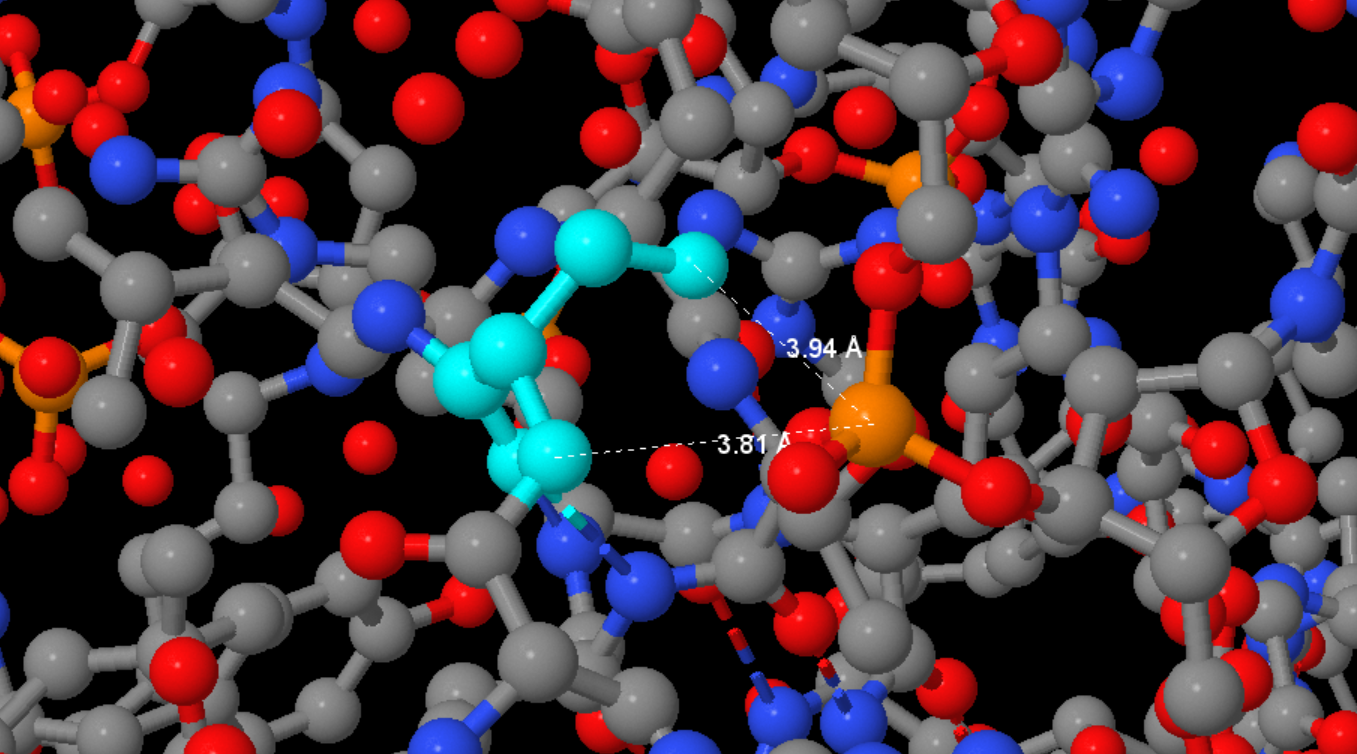
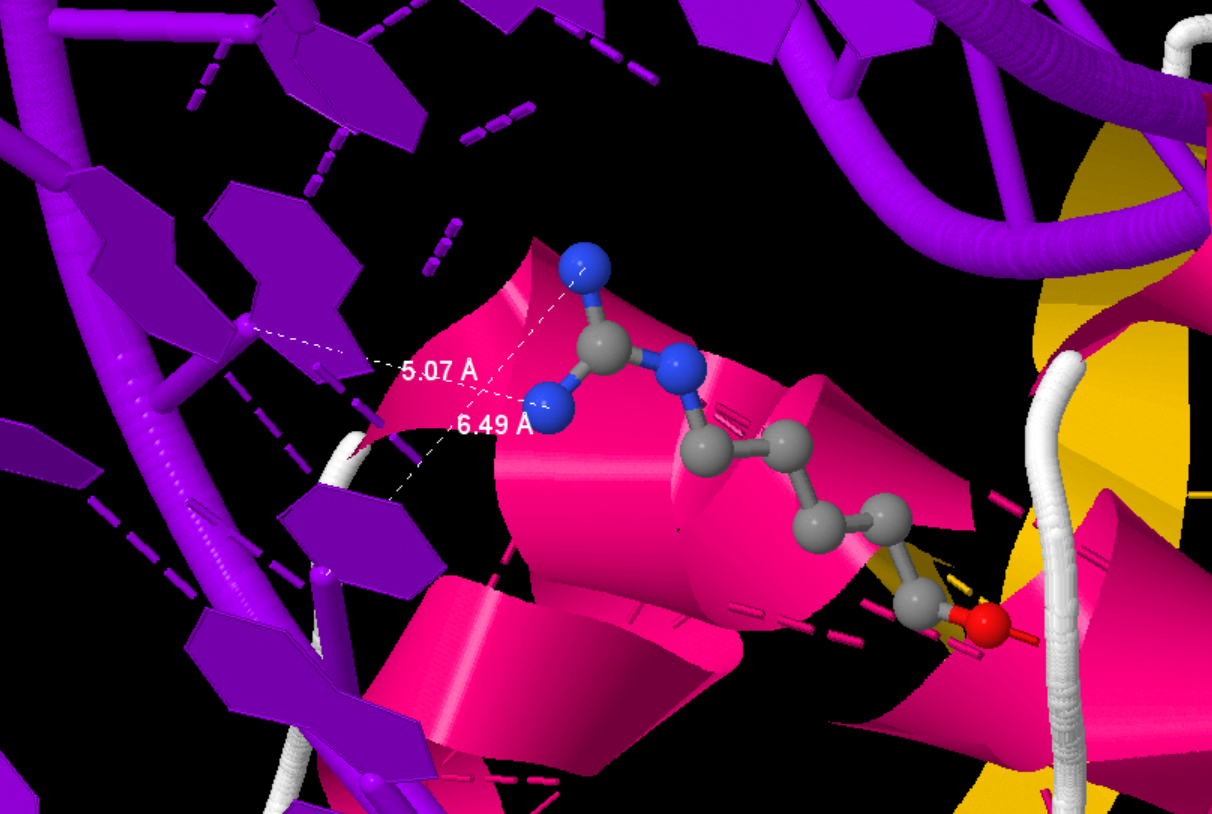
Searching for DNA-protein contacts in a given structure
Reference to a script highlighting the oxygen atoms of 2'-deoxyribose, phosphoric acid residues, and nitrogen atoms of nitrogenous base residues in JMol.
Reference to a script whose call to JMol will give a sequential image of the whole structure, only DNA in a wire model, the same model, but with the selected beads set1, then set2 and set3 atoms.
We will consider the oxygen and nitrogen atoms as polar and the carbon, phosphorus, and sulfur atoms as non-polar. We will call as polar contact the situation in which the distance between the polar protein atom and the polar DNA atom is less than 3.5 Å. Similarly, we will consider a pair of non-polar atoms at a distance less than 4.5 Å as a non-polar contact. Below is a table comparing the number of contacts of different types:
| Contacts of protein atoms with | Polar | Non-polar | Total |
|---|---|---|---|
| 2'-deoxyribose residues | 4 | 22 | 26 |
| phosphoric acid residue | 10 | 12 | 22 |
| residual nitrogenous bases on the side of the large furrow | 3 | 5 | 8 |
| residual nitrogenous bases on the side of the small furrow | 1 | 0 | 1 |
Obtaining a DNA-protein contact diagram using the nucplot program
link to the .ps file
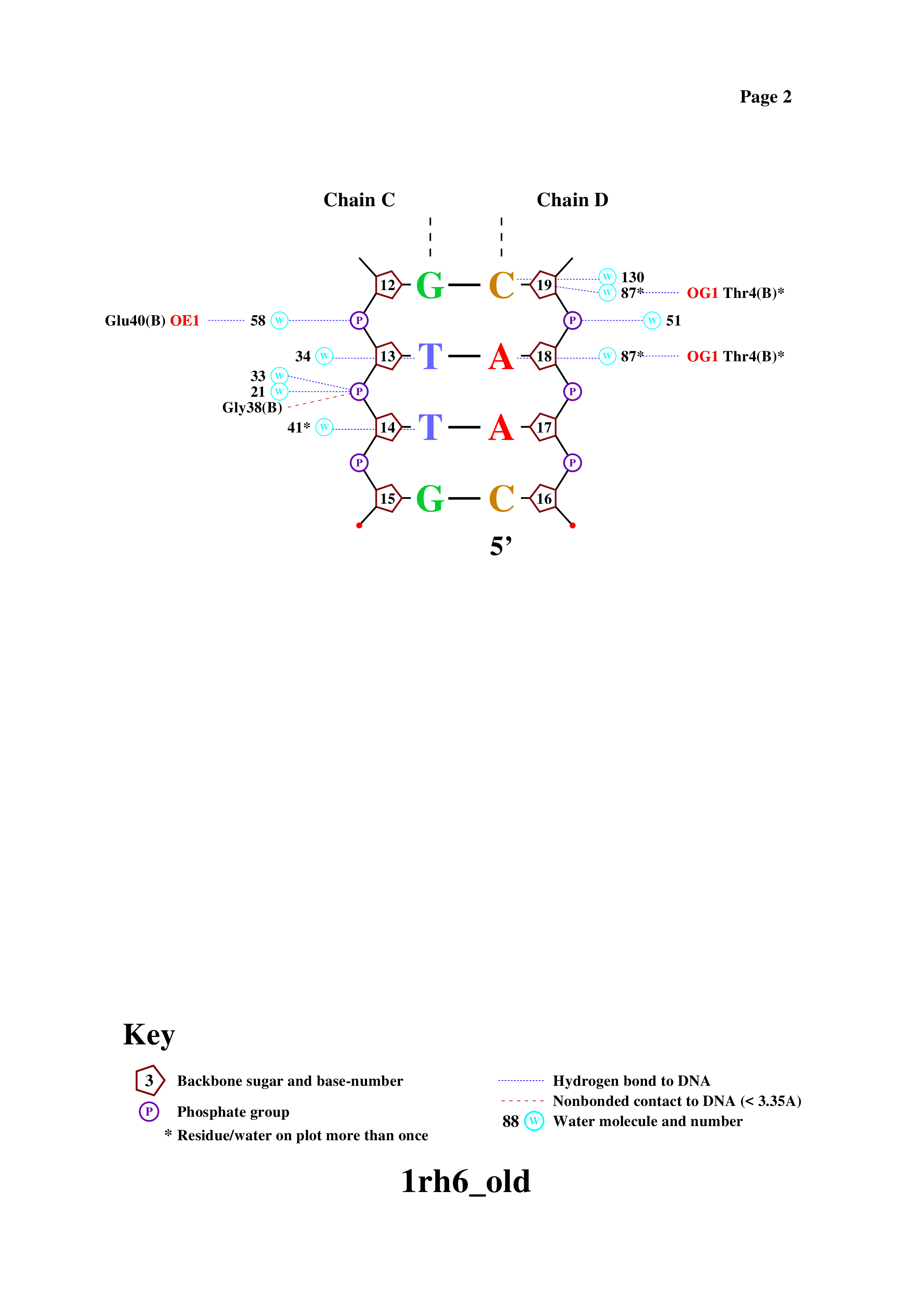
Based on this scheme were selected:
The amino acid residue with the greatest number of contacts with DNA shown in the diagram is Ser17(B) (2 contacts)
The amino acid residue most important for DNA sequence recognition is Arg23(B), which is bound to two nitrogenous base residues, not to the DNA backbone