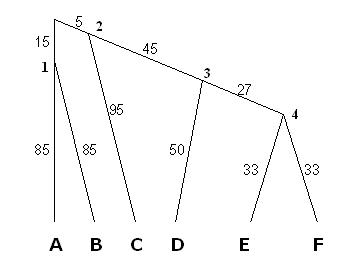
Сначала целесообразно продемонстрировать то дерево, которое было исходным.
1) Первым этапом нашей работы явилось выравнивание
последовательностей, соответствующих листьям дерева (A, ..., F).
Последовательности в формате fasta были записаны в один файл (summa.fasta).
Программой EMBOSS это воспринимается как выравнивание.
2) С помощью программы ednadist была построена квадратная матрица попарных эволюционных расстояний по методу Джукса-Кантора. Вот она.
|
В этой матрице показаны попарные расстояния между последовательностями A, ..., F. Матрица квадратная, поэтому элемент (i, j) (где i — строка, j — столбец) совпадает по значению с элементом (j, i). |
3) Теперь рассмотрим 4 филогенетических дерева, которые были получены с
помощью разных программ.
а) Метод UPGMA, программа eneighbor.
+----------------------------------A
+-------3
+--4 +----------------------------------B
! !
! +------------------------------------------C
--5
! +-------------E
! +---------1
+--------------------2 +-------------F
!
+-----------------------D
Здесь 1-5 — номера узлов (не совпадают с теми номерами, которые были на
исходном дереве), A, ..., F — обозначения листьев.б) Метод Neigbor-Joining (ближайших соседей), программа eneighbor.
+-------------------------------------------C
!
! +-----------E
! +--------------1
--4---------------------2 +---------------F
! !
! +----------------D
!
! +---------------------------A
+------3
+----------------------------------------B
Обозначения те же.в) Метод Maximum Likelihood (наибольшего правдоподобия), программа ednaml.
+----------D
+----------------4
! ! +--------F
+-----2 +--------3
! ! +------E
! !
! +------------------------------C
!
--1---------------------------B
!
+--------------------A
Обозначения те же.г) Метод Parsimony (максимальной экономии), программа ednapars.
+-----------B
!
+--2 +-----D
! ! +--5
! ! ! ! +--F
! +--3 +--4
--1 ! +--E
! !
! +--------C
!
+--------------A
Обозначения те же.4) Теперь сравним топологию полученных деревьев. Для этого составим следующую таблицу:
|
Исходное дерево модели | UPGMA | NJ | ML | Parsimony | ||
110000 |
+ | + | + | + | + | ||
111000 |
+ | + | + | + | + | ||
111100 |
+ | + | + | + | + |
5) Наконец, опишем результаты сравнения деревьев.
Из таблицы видно, что внутренние ветви всех деревьев совпадают. Это говорит о
том, что реконструкция хода эволюции достаточно надёжна, причём всеми методами.
Однако метод UPGMA, как мне представляется, более эффективен (то есть
полученное дерево ближе всего к исходному). Почему?
Дерево является ультраметрическим, а исходное — почти ультраметрическим
(расстояние до 4 из 6 ветвей равно 100, а до двух — 110).
Пожалуй, стоит ещё заметить, что лишь дерево, построенное по методу UPGMA,
является укоренённым. Это даёт ещё больше оснований доверять данному методу
(для восстановления процесса эволюции). Ультраметрическое же дерево
получилось лишь в первом случае (опять-таки метод UPGMA ;).
И в завершение: первые два рассматриваемых метода (UPGMA и Neigbor-Joining)
используют матрицу попарных расстояний, а два других — выравнивание
последовательностей (т.е. являются символьно-ориентировочными).