Гомологичное моделирование комплекса белка с лигандом
Цель данного занятия - ознакомиться с возможностями гомологичного моделирования комплекса белка с лигандом.
Мой белок LYS_CLOAB - лизоцим из Clostridium acetobutylicum. В качестве образца была использована известная структура лизоцима из форели (1lmp).
Входные данные использованные программой MODELLER для моделирования структуры белка :
Было построено выравнивание последовательности из структуры лизоцима форели с последовательностью белка LYS_CLOAB с помощью Clustal и сохранено в формате PIR. Затем полученное выравнивание было модифицированно следующим образом:
-была переименована последовательность в файле:
| Было | Стало |
>P1;sp|P34020|LYS_CLOAB |
>P1;seq |
>P1;1LMP_A|PDBID|CHAIN|SEQUENCE |
>P1;1lmp |
-после имени последовательности моделируемого белка была добавлена строчка,описывающая входные параметры последовательности для modeller:
sequence:ХХХХХ::::::: 0.00: 0.00-после имени последовательности белка-образца была добавлена строчка, описывающая, какой файл содержит структуру белка с этой последовательностью, номера первой и последней аминокислот ( 130 а не 132 !!!) в структуре,идентификатор цепи и т.д.:
structureX:1lmp_now.ent:1 :A: 130 :A:undefined:undefined:-1.00:-1.00-в конце каждой последовательности была добавлена точка (/ не нужен!!!), которая указывает на то, что имеется один лиганд (если бы было два лиганда стояли бы две точки).
Был скачен файл в формате pdb ,затем была удалена вся вода из структуры (строчки типа: HETATM xxxx O HOH ...), всем атомам лиганда присвоен один и тот же номер "остатка" (MODELLER считает, что один лиганд = один остаток) и модифицированы имена атомов каждого остатка за счет добавления в конец буквы A, B, C (Смысл операции в том, что атомы остатка 130 имели индекс А, атомы остатка 131 имели индекс В и т.д.), после чего номера остатков были изменены на 130 ( исключение: у атома O1L ничего не добавляли, чтобы не было слишком много букв). Заменили NDG на NAG и сохранили все в файле 1lmp_now.ent
В скрипте были удалены ":A" при указании контакта, а также были отредактированы строки, в которых указаны какие водородные связи белка с лигандом должны быть в будущей модели. Номера остатков и имена нужных атомов были определены по выравниванию и тому, какие водородные связи имеются в образце(критерий водородной связи: расстояние менее 3.5 ангстрем между азотом или кислородом белка с подходящими атомами лиганда).
Поиск водородных связей в образце:
select NAG , resi 130 or resi 131 or resi 132 select water , /1LMP//A/HOH select None select (all within 3 of NAG) and not water
Результат поиска водородных связей ( атомы образующие водородную связь: в образеце/ в лиганде/ в будущей модели):
1LMP NAG SEQ TYR 62 CE2 NAG 130 O6B ALA 155 CB -не использовала ASP 52 OD2 NAG 130 O1L GLU 145 OE2 ASN 59 N NAG 130 O7B ASN 152 C ASP 101 OD2 NAG 130 O6A ASP 193 OD2Т.к. в моделируемом белке число остатков не совпадает с числом остатков в белке-образце, то номера "остатков" лиганда были тоже изменены на № последнего остатка+1=325.
Полученный скрипт был запущен командой:
mod9v7 LYS_CLOAB.pyРезультат:
модель №1
модель №2
модель №3
модель №4
модель №5
Анализ результатов
Сравнение моделей визуально:Все модели отличаются положением вариабельных петель, при этом все а-спирали и b-листы практически полностью совпадают. Но при этом складывается такое впечатление,что часть белка вообще не уложена (не только концы, но и часть в середине). Что вполне логично, т.к. исследуемый белок значительно больше образца.
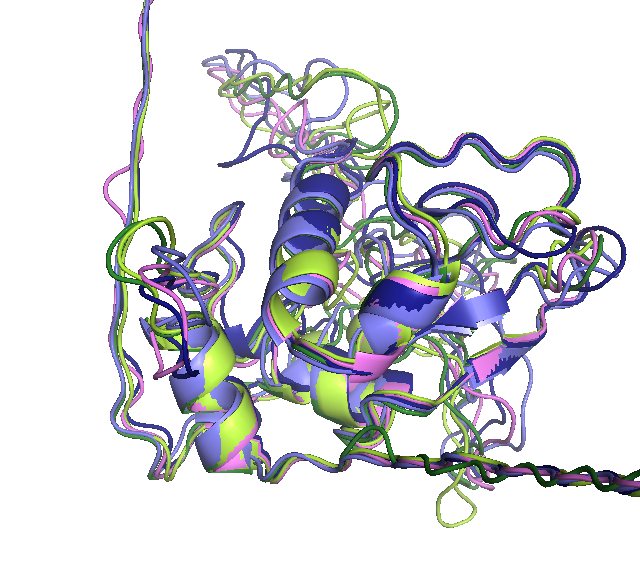 |
№1 - голубой №2 - лимонный №3 - синий №4 - фиолетовый №5 - зеленый |
Проверка качества моделей и выбор лучшей:
Использванные инструменты для оценки качества структуры - WHATIF (Structure validation).
№ model Number of anomalous Number of anomalous
bond lengths(Z-score) bond angles (Z-score)
1 2 (0.967) 91 (1.393)
2 2 (0.966) 82 (1.390)
3 5 (0.963) 80 (1.371)
4 1 (0.952) 86 (1.394)
5 2 (0.973) 79 (1.391)
Лучшей моделью, на мой взгляд, является №5, т.к. она имеет самое маленькое число аномальных углов с хорошим Z-score и небольшое число
аномальных связей с самым близким к 1 значением Z-score.
Для моделирования с помощью образца все-таки лучше использовать белки примерно одинаковой длины, чтобы не было таких вот прямых хвостов.