Докинг низкомолекулярных лигандов в структуру белка
Цель данного занятия - ознакомиться с возможностями докинга низкомолекулярного лиганда в структуру белка.
Используемый пакет - Autodock Vina (входные данные - специально форматированные файлы pdb c зарядами и указанием торсионных углов) и Autodock tools.
Была взята модель №5 белка лизоцима, построенная на основе гомологичного моделирования на прошлом практикуме, и из нее был удален лиганд.
Докинг одного из мономеров сахара (NAG) из прошлого занятия
В банке pdb была найдена SMILES нотация для NAG и сохранена в файл nag.smi :CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O
C помощью obgen была построена 3D структуру этого сахара в pdb формате :
obgen nag.smi > nag.mol babel nag.mol nag.pdb
Скриптом prepare_ligand4.py из пакета Autodock tools был создан pdbqt файл данного лиганда:
export PATH=${PATH}:/home/preps/golovin/progs/bin
prepare_ligand4.py -l nag.pdb
Так же, скриптом prepare_receptor4.py из пакета Autodock tools был создан pdbqt файл модели №5 лизоцима.
prepare_receptor4.py -r seq.B99990005.pdb
Затем был создан файл с параметрами докинга vina.cfg . Для докинга была указана область поиска места связывания в структуре белка - куб с центором, координаты которого были определены с помощью PyMol из модели №5 комплекса, как координаты центра масс между остатками, образующими водородные связи с легандом(№ остатков взяты из предыдущего практикума):
pseudoatom NAG, resi 145+152+193
Координаты центра можно посмотреть, если выделить его, а затем сохранить как молекулу в отдельный файл ,выбрав нужное выделение.
Далее был проведен первый докинг( результат - nag_prot.pdbqt, nag_prot.log ):
vina --config vina.cfg --receptor seq.B99990005.pdbqt --ligand nag.pdbqt --out nag_prot.pdbqt --log nag_prot.log
Энергии 3ёх лучших расположений и геометрическая разница между ними:
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.1 0.000 0.000
2 -4.9 1.461 2.521
3 -4.7 1.759 2.008
* rmsd u.b. - среднеквадратичное отклонение, не учитывающее симметрию (нам нужно именно оно)
Подсчет геометрической разницы:
между 1 и 2 : 0.000 - 2.521 = -2.521
между 1 и 3 : 0.000 - 2.008 = -2.008
Резудьтат загрузки в PyMol файлов nag_prot.pdbqt и seq.B99990005.pdbqt (отображены все состояния лиганда):
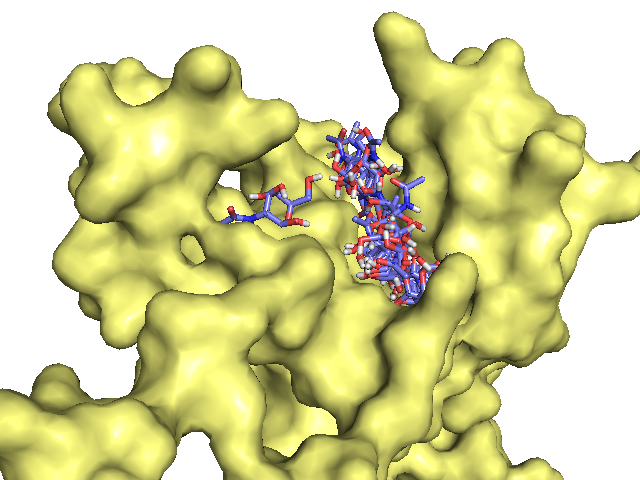
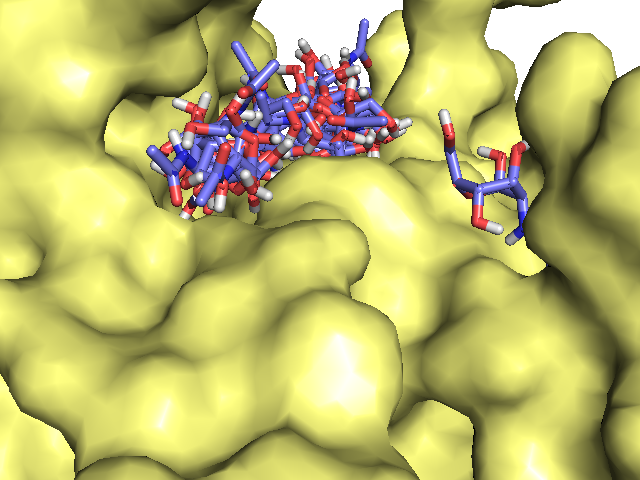
Затем был проведен докинг с рассмотрением подвижности некоторых боковых радикалов белка. Сначала белок был разбит на две части, подвижную и неподвижную . Для подвижной части были выбраны 3 аминокислоты, использованные в прошлом задании для позиционирования лиганда:
python /usr/share/pyshared/AutoDockTools/Utilities24/prepare_flexreceptor4.py -r seq.B99990005.pdbqt -s GLU145_ASN152_ASP193и проведен докинг ( результат - nag_prot_flex.pdbqt, nag_prot_flex.log ):
vina --config vina.cfg --receptor seq.B99990005_rigid.pdbqt --flex seq.B99990005_flex.pdbqt --ligand nag.pdbqt --out nag_prot_flex.pdbqt --log nag_prot_flex.log
Энергии 3ёх лучших расположений и геометрическая разница между ними:
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.0 0.000 0.000
2 -4.9 2.028 3.297
3 -4.8 2.000 3.426
* rmsd u.b. - среднеквадратичное отклонение, не учитывающее симметрию (нам нужно именно оно)
Подсчет геометрической разницы:
между 1 и 2 : 0.000 - 3.297 = -3.297
между 1 и 3 : 0.000 - 3.426 = -3.426
Интересно отметить, что при перезапуске скрипта значения энергии и rmsd меняются.По времени докинг с подвижными радикалами происходит значительно дольше,чем обычный.
Резудьтат загрузки в PyMol файлов nag_prot_flex.pdbqt и seq.B99990005_rigid.pdbqt (отображены все состояния лиганда):
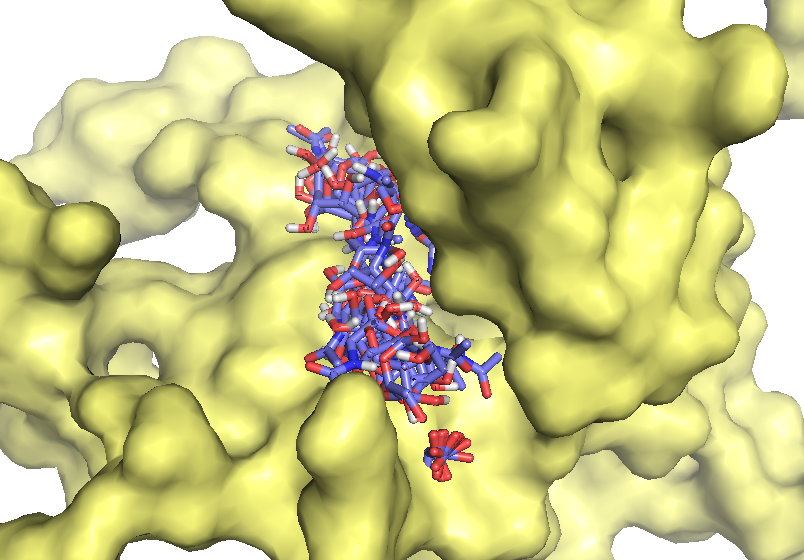
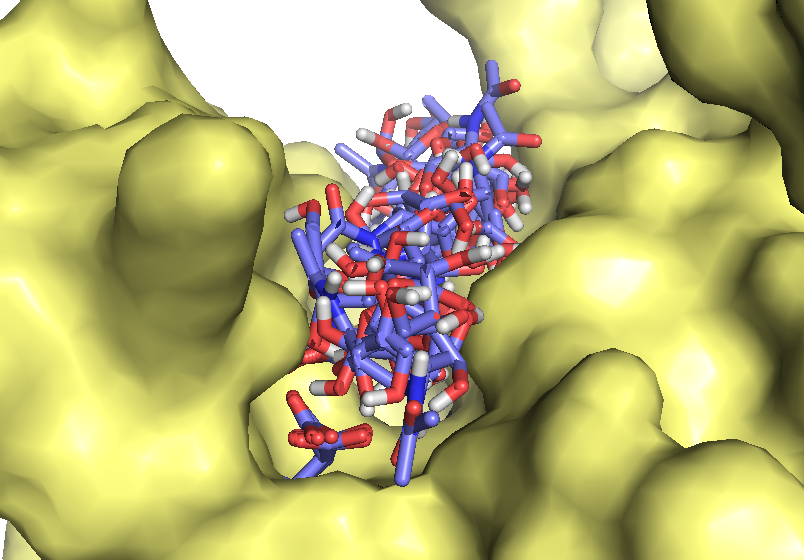
######В отчёт надо занести различия которые Вы обнаружите по сравнению с обычным докингом.
######Сделайте вывод, может ли докинг расположить лиганд наиболее близким образом к тому, что вы получили в моделировании. Если да, то отметьте энергетическую эффективность этого расположения. Прикрепить картинку : докинг+ лиганд из прошлого занятия
Далее были созданы 4 лиганда, где метильный радикал группы СH3C(=O)NH из NAG был заменён на одну из следующих групп(OH, NH2, H, Ph),после чего для каждого из этих лигандов был проведен обыкновенный докинг и докинг с подвижными радикалами (кроме Н):
| замена | SMILES | 3D стр-ра (pdb) | pdbqt файл | обычный докинг | докинг с подвижными радикалами |
| OH | nag_OH.smi | nag_OH.pdb | nag_OH.pdbqt | nag_OH_prot.log
nag_OH_prot.pdbqt |
nag_OH_prot_flex.log
nag_OH_prot_flex.pdbqt |
| NH2 | nag_NH2.smi | nag_NH2.pdb | nag_NH2.pdbqt | nag_NH2_prot.log
nag_NH2_prot.pdbqt |
nag_NH2_prot_flex.log
nag_NH2_prot_flex.pdbqt |
| H | nag_H.smi | nag_H.pdb | nag_H.pdbqt | nag_H_prot.log
nag_H_prot.pdbqt |
_____
_____ |
| Ph | nag_Ph.smi | nag_Ph.pdb | nag_Ph.pdbqt | nag_Ph_prot.log
nag_Ph_prot.pdbqt |
nag_Ph_prot_flex.log
nag_Ph_prot_flex.pdbqt |
OH 1 -5.2 0.000 0.000 2 -5.1 8.548 9.886 3 -4.8 2.441 4.246 1 -5.1 0.000 0.000 2 -4.8 1.414 2.329 3 -4.6 4.191 7.879 NH2 1 -5.3 0.000 0.000 2 -5.0 2.813 4.171 3 -4.9 1.792 2.753 1 -5.3 0.000 0.000 2 -5.0 2.375 3.177 3 -4.9 1.558 2.271 H 1 -4.5 0.000 0.000 2 -4.4 2.470 4.087 3 -4.4 1.409 1.434 --- Ph 1 -6.9 0.000 0.000 2 -6.5 1.995 2.511 3 -6.5 1.407 2.539 1 -6.6 0.000 0.000 2 -6.6 2.432 3.723 3 -6.5 2.496 4.343