| Командная строка | Функция |
cp chr16.2.fastq /nfs/srv/databases/ngs/fp.delta/pr12 |
Копиравание файлов с одноконцевыми чтениями в формате fastq |
fastqc chr16.2.fastq |
Обработка FastQC |
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr16.fastq chr16_trim.fastq TRAILING:20 MINLEN:50 |
Обрезание концов |
hisat2-build ../chr16.fasta indexed |
Индексирование |
hisat2 -x indexed -U chr16.2_trim.fastq -S chr16.2_aligntoref.sam --no-softclip |
Картирование чтений из fastq (по индексированной последовательности) |
samtools view -b chr16.2_aligntoref.sam -o chr16.2_align.bam |
Конвертнация в .bam |
samtools sort chr16.2_align.bam chr16_align_sorted |
Сортировка по координате в начале чтения |
samtools index chr16.2_align_sorted.bam |
Индексация отсортированного .bam |
htseq-count -i gene_id -s no -m union -f bam chr16.2_align_sorted.bam /nfs/srv/databases/ngs/Human/rnaseq_reads/gencode.v19.chr_patch_hapl_scaff.annotation.gtf -o count.sam >> ready.txt |
Подсчёт ридов |
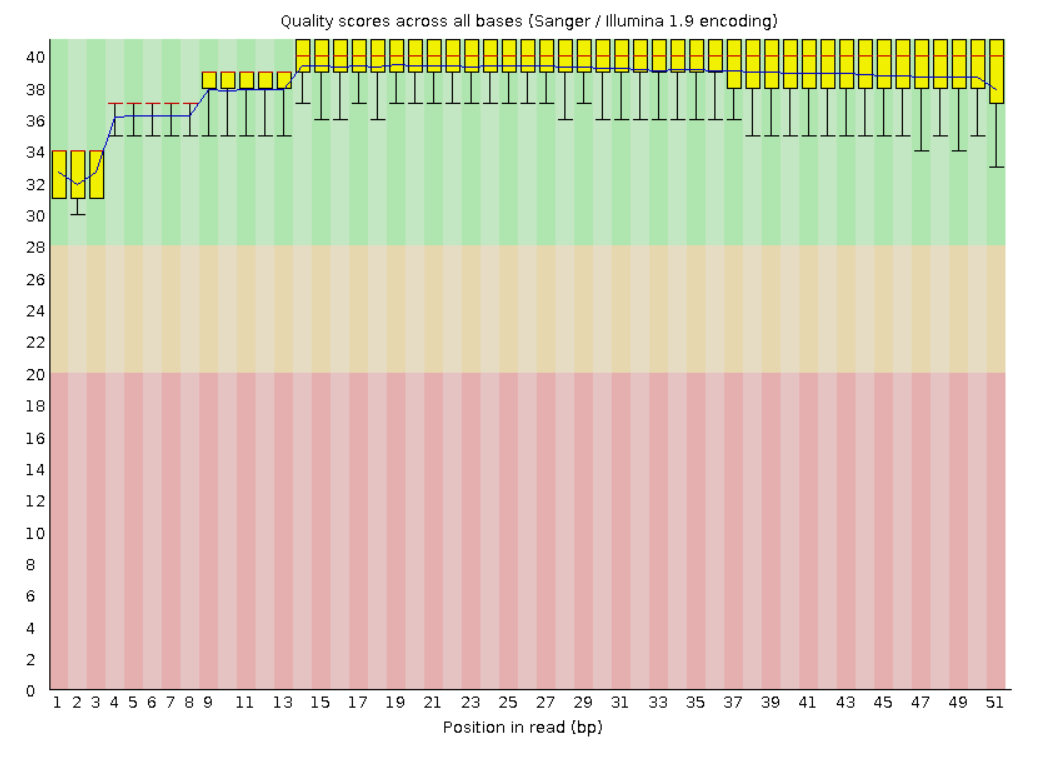
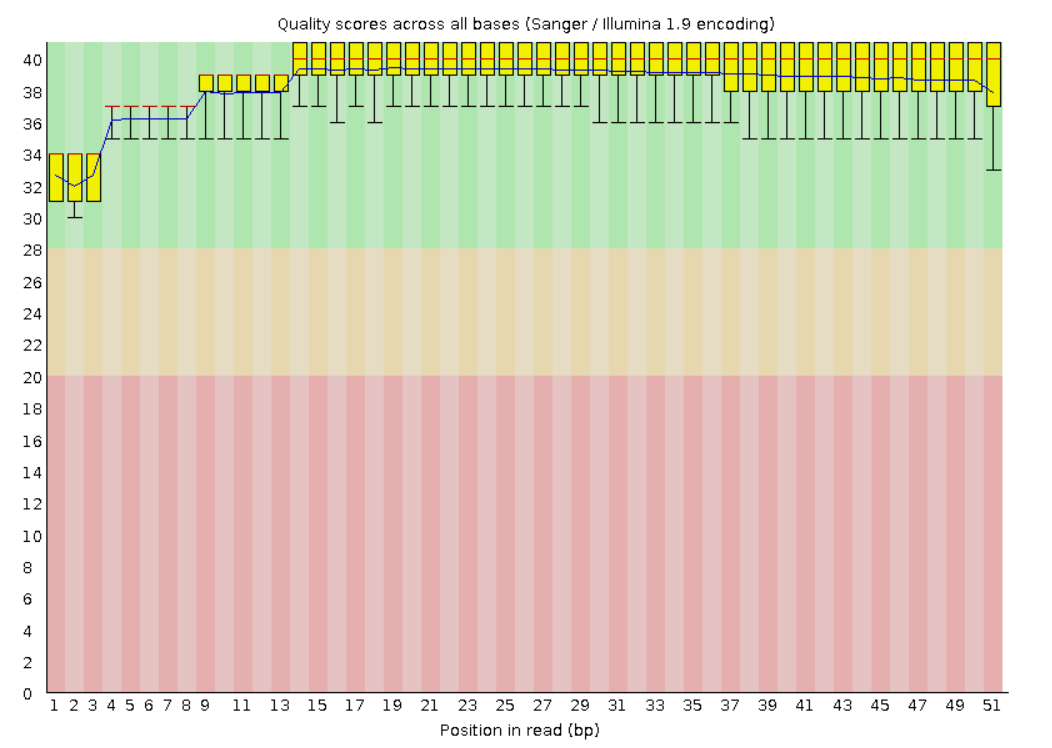
рафики качества чтения оснований были сгненерированы fastqc
На этих графиках красной линий обозначается медиана, желтый блок означает межквартильный диапазон, верхние и нижние усы - 10 и 90%, синяя линия - среднее значение
По оси y отложены quality scores
До обрезки:
После обрезки:
Результаты:
Input Reads: 9517 Surviving: 9469 (99,50%) Dropped: 48 (0,50%)
В результате ~99% сохранились, что не особо отличаетя от изначальных данных
98.41% чтений были картированы на хромосому, качество можно считать довольно высоким
9469 reads; of these:
9469 (100.00%) were unpaired; of these:
151 (1.59%) aligned 0 times
9318 (98.41%) aligned exactly 1 time
0 (0.00%) aligned >1 times
98.41% overall alignment rate
__no_feature 3809 __ambiguous 0 __too_low_aQual 0 __not_aligned 151 __alignment_not_unique 0
В результате получили 5509 чтений ENSG00000166501.8
Выдача NCBI по запросу: