В банке pdb найдите SMILES нотацию для NAG (на странице 1lmp) и создадим в файл nag.smi.
Далее, с помощью obgen строим 3D структуру этого лиганда в pdb формате:
obgen nag.smi > nag.mol
babel -imol nag.mol -opdb nag.pdb
Теперь, скриптом prepare_ligand4.py из пакета Autodock tools создаем nag.pdbqt файл лиганда:
export PATH=${PATH}:/home/preps/golovin/progs/bin
prepare_ligand4.py -l nag.pdb
Скриптом prepare_receptor4.py из пакета Autodock tools создаем protein.pdbqt файл белка. Я брал четвертую модель из предыдущего задания!
prepare_receptor4.py -r seq.B99990004.pdb -o protein.pdbqt
Далее, я создал файл vina.cfg с таким содержанием (pseudoatom определил центр масс где-то в глубине белка, но ближайшим к нему атомом оказался один из атомов лиганда, его координаты и были взяты за основу):
center_x=40.523
center_y=39.883
center_z=26.170
size_x = 25
size_y = 25
size_z = 25
num_modes = 20
Теперь можно провести первый докинг!
vina --config vina.cfg --receptor prot.pdbqt --ligand nag.pdbqt --out nag_prot.pdbqt --log nag_prot.log
В файле nag_prot.log можно посмотреть на энергии 3ёх лучших расположений и геометрическую разницу между ними:
mode | affinity | dist from
| (kcal/mol) | rmsd l.b.
-----+------------+----------+
1 -5.2 0.000
2 -5.1 2.401
3 -4.6 1.921
На картинке изображены все состояния лиганда (видно, что большинство из них занимает относительно правильное положение, но есть и "выбросы"!):
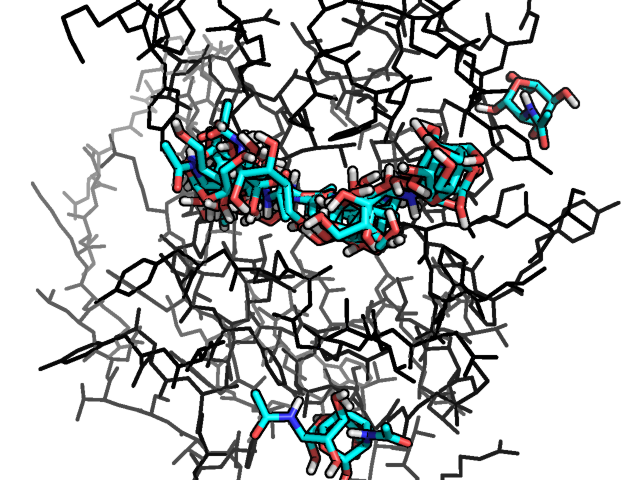
Теперь давайте проведём докинг рассматривая подвижность некоторых боковых радикалов белка. Сначала разобьем белок на две части, подвижную и неподвижную. Для подвижной части выберем 3 аминокислоты которые мы использовали в прошлом задании для позиционирования лиганда. А затем проведем докинг!
python /usr/share/pyshared/AutoDockTools/Utilities24/prepare_flexreceptor4.py -r prot.pdbqt -s CYS54_ALA103_TYR105
vina --config vina.cfg --receptor protein_rigid.pdbqt --flex protein_flex.pdbqt --ligand nag.pdbqt --out nag_prot_flex.pdbqt --log nag_prot_flex.log
В файле nag_prot_flex.log можно посмотреть на энергии 3ёх лучших расположений и геометрическую разницу между ними:
mode | affinity | dist from
| (kcal/mol) | rmsd l.b.
-----+------------+----------+
1 -5.2 0.000
2 -5.1 3.092
3 -4.9 1.767
И привожу соответствующее изображение со всеми состояниями лиганда:
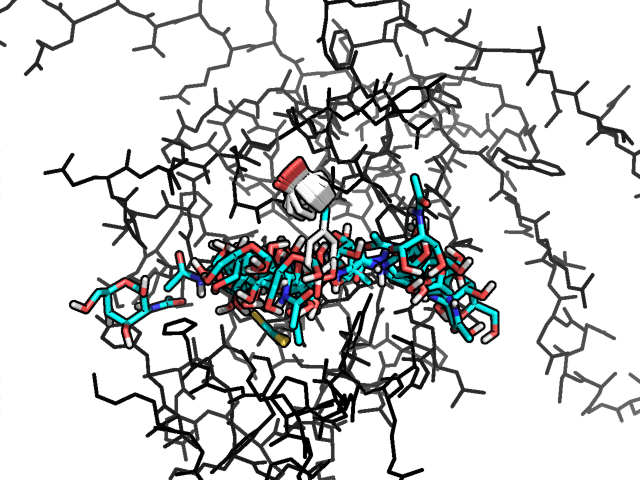
Может ли докинг расположить лиганд наиболее близким образом к тому, что вы получили в моделировании? Да, может, и энергетическая эффективность этого довольно велика (можно посмотреть на соответствующие значения энергии сродства, которые приведены выше). Более того, при докинге мы можем моделировать подвижность аминокислот в нашем белке. А это более биологически осмысленно, поскольку при связывании лиганда аминокислоты белка также, как правило, двигаются.
NAG содержит в себе СH3C(=O)NH группу. Создадим 4 лиганда где метильный радикал этой группы будет заменён на OH, NH2, H, Ph. Для каждого из этих лигандов проведем обыкновенный докинг (и докинг с подвижными аминокислотами в белке) и представим результаты в виде таблицы из трёх лучших расположений для каждого лиганда. Были получены следующие файлы: nag2.smi, nag3.smi, nag4.smi и nag5.smi. В каждом из них был заменен метильный радикал на соответствующую группу. Для каждого из них проводим процедуры аналогичные тем, что были сделаны выше. Для сравнения привожу результаты докинга для лиганда с фенильной группой (для неподвижных радикалов - слева, для подвижного - справа):
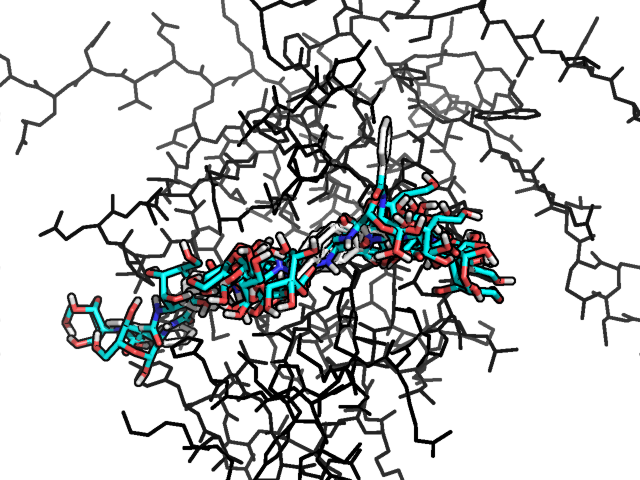
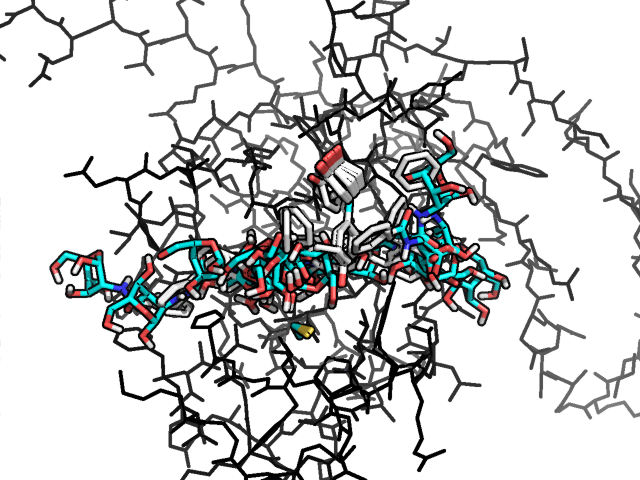
|
|
OH
|
NH2
|
H
|
Ph
|
|
1
|
-5.1
|
-5.2
|
-4.7
|
-6.2
|
|
2
|
-4.9
|
-5.1
|
-4.6
|
-5.9
|
|
3
|
-4.7
|
-5.0
|
-4.5
|
-5.8
|
|
|
OH
|
NH2
|
Ph
|
|
1
|
-5.1
|
-5.2
|
-6.1
|
|
2
|
-4.9
|
-5.1
|
-6.1
|
|
3
|
-4.6
|
-4.9
|
-5.9
|