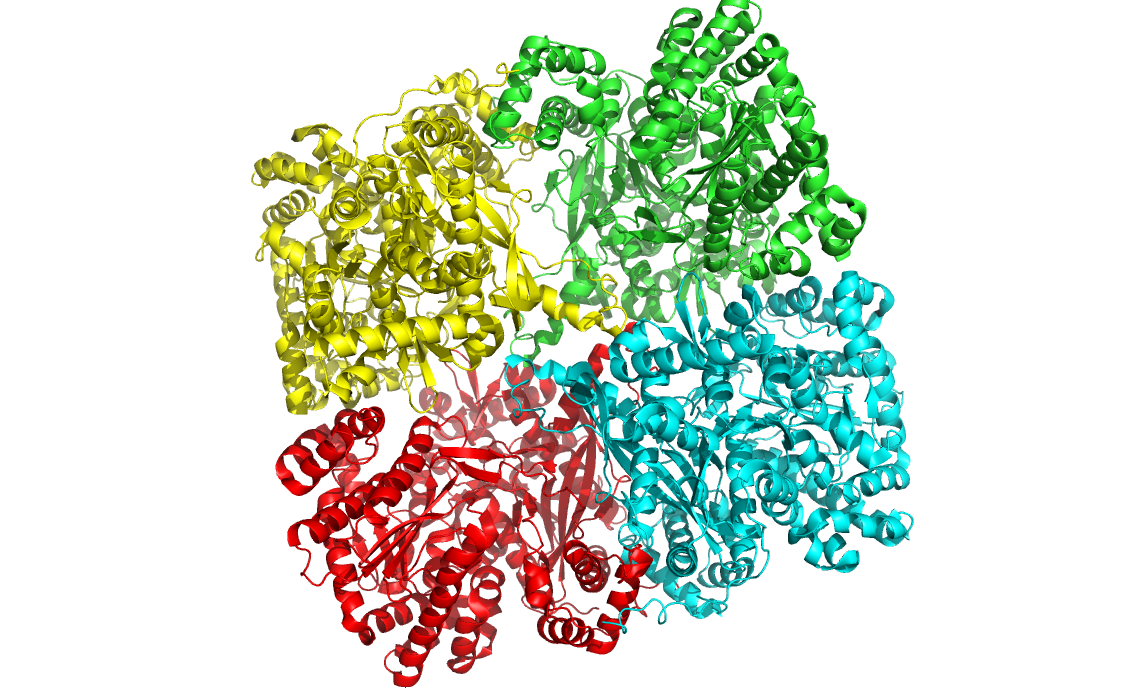
№1 – 7GPB
Год расшифровки: судя по всему 1991, так как статья (Structural mechanism for glycogen phosphorylase control by phosphorylation and AMP), в которой представлена данная структура, была опубликована в журнале J.Mol.Biol именно в этом году (но в самом PDB файле упоминается 1990 год).
Разрешение: 2.90 ангстрем (удовлетворительное).
Странности: Сразу напишу, что мне не понравилось в данном PDB файле. Раздел DATA USED IN REFINEMENT. Тут указано разрешение самого лучшего рефлекса (2.90 ангстрем). А дальше по нулям. Нет разрешения худшего рефлекса, нет количества рефлексов, нет полноты данных. Или в начале девяностых было непринято такие данные класть в PDB файл? R-factor равен 0.171, что неплохо, однако, нет значения R-free. Это как раз понятно, так как считать R-free стали после статьи Клевегта (Kleywegt) 1995 года. Проблема в этой структуре заключается в триптофанах. Если внимательно рассмотреть каждый из триптофанов, то можно увидеть, что с ними не так. В боковой группе кольца находятся под углом относительно друг друга! Они не лежат в одной плоскости, что странно. Привожу самые яркие примеры подобных несуразиц.
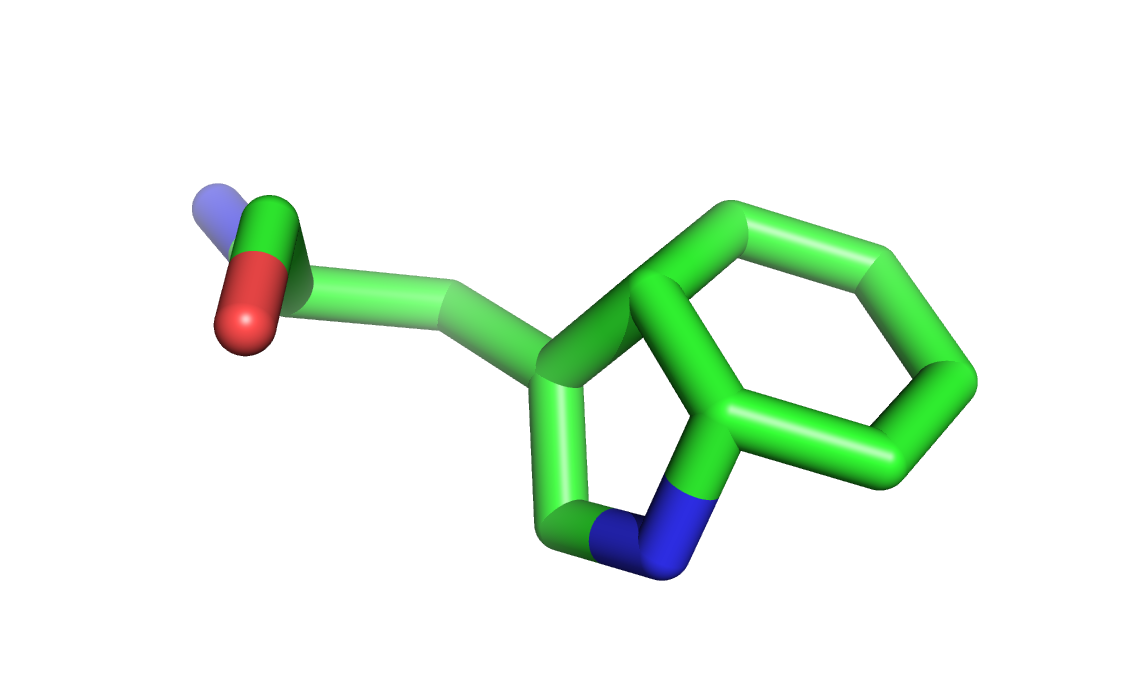
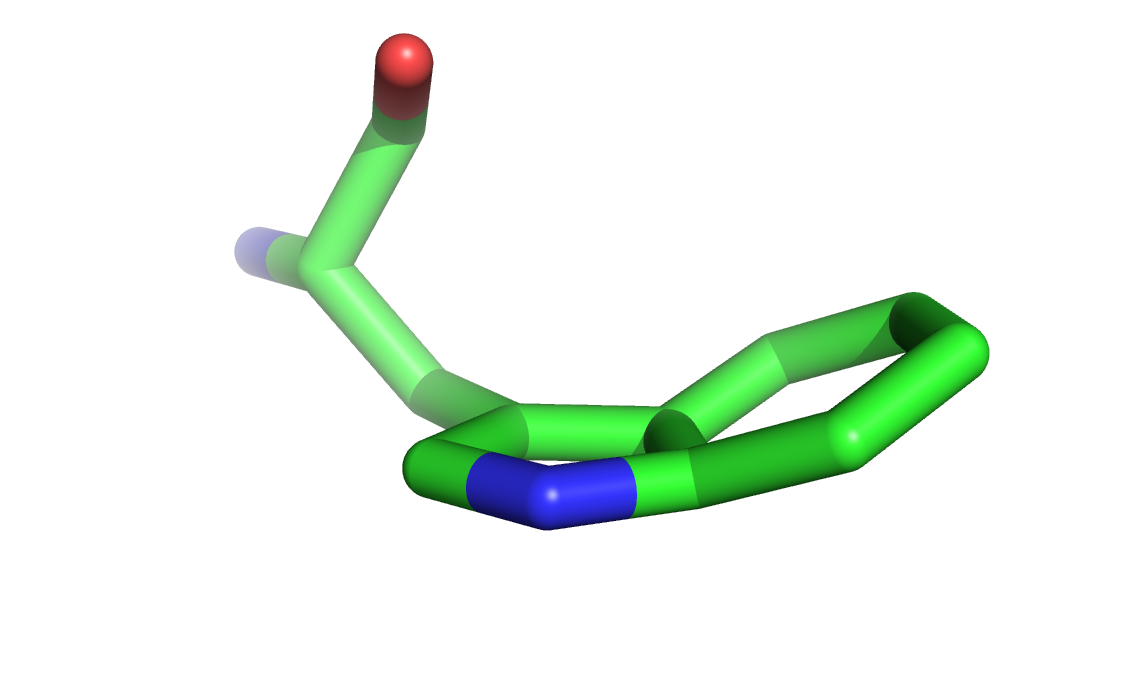
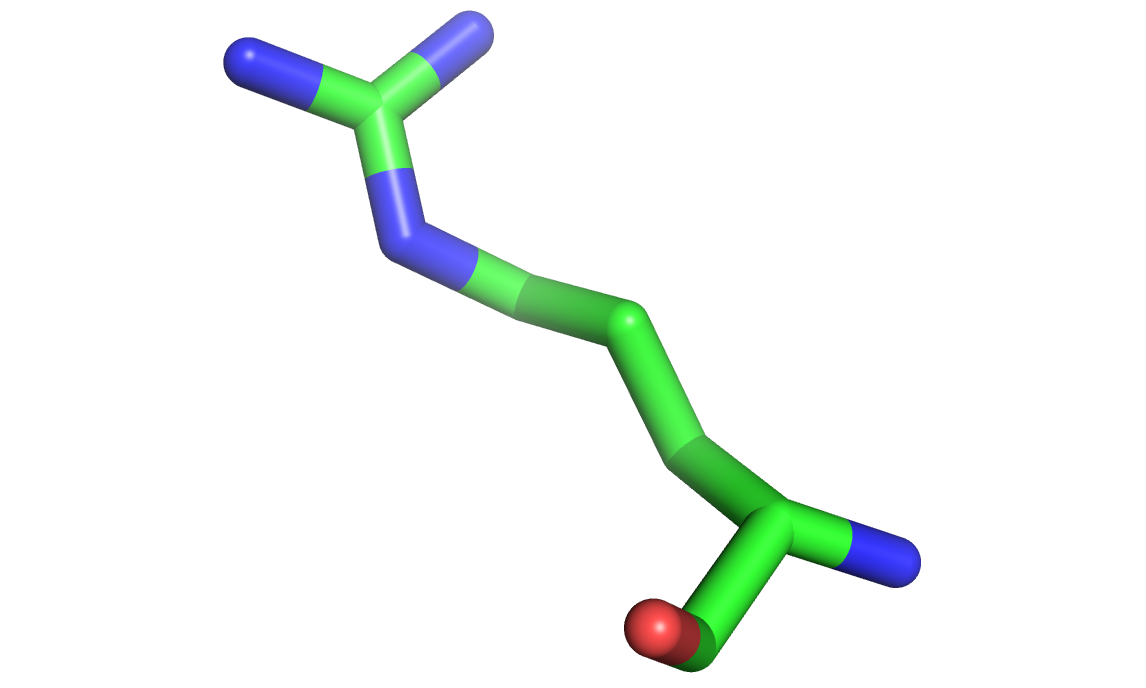
Слева изображен 67 триптофан цепи D, справа – триптофан 67 цепи A. В чем причина? Я не знаю точно. Наверное, при таком нехорошем разрешении авторам не удалось правильно вписать модель в полученную электронную плотность, или сама модель определена не очень хорошо (без R-free сложно судить). Вначале я думал, что дело в температурных факторах, потому что они сильно скачут у этих неправильных триптофанов (доходит до 50), однако, есть неправильные триптофаны, у которых температурный фактор вполне нормальный (например, триптофан 361 из цепи В). Так что, сказать точно здесь нельзя. Помимо этого, хочется обратить внимание на остаток аргинина 184 цепи А – у него есть атомы с очень высокими температурными факторами, однако, со структурой вроде все нормально. Ниже представлена структура этого самого аргинина.
Теперь я возьму на себя смелость перечислить номера всех триптофанов, с которыми, на мой взгляд, не совсем все в порядке: chain A (67, 189, 215, 491, 825), chain B (182, 361, 387, 491, 797), chain C (67, 244, 365, 387), chain D (67, 797).
№2 – 1GT0
_1.png)
Год расшифровки: статья - Crystal structure of a POU/HMG/DNA ternary complex suggests differential assembly of Oct4 and Sox2 on two enhancers. 2003 год. Журнал - Genes
Разрешение: 2.60 ангстрем (удовлетворительное). В структуре присутствуют молекулы воды, однако условное правило гласит, что молекулы воды в структуру вносятся только при условии, что разрешение лучше 2.50 ангстрем.
Странности: В этой структуре лучший рефлекс имеет разрешение 2.60 ангстрем, худший – 30 ангстрем. Полнота составляет 98.2%, и это является хорошим показателем. Однако, R-factor равен 23.2%, что нельзя назвать ни хорошим, ни плохим значением. Подсчитан R-free, он равен 28.5%, что тоже ни хорошо, ни плохо. Конечно же, первое, что бросается в глаза в этой структуре, это очень странное расположение одного из концевых участков ДНК (на рисунке выше выделено красным). Но главная странность – отсутствие с 78 по 96 включительно аминокислот в цепи C. На рисунке ниже это изображено. Желтая цепь – это остатки от 101 до 158 в цепи C. Синяя цепь – остатки от 1 до 74 в цепи С. Красным помечены остатки 100-97 и 75-77. Показан только остов полипептидной цепи, боковые группы удалены.
_2.png)
С чем это может быть связано? Это можно было бы объяснить occupancy, но остатки 75-77 и 97-100 представлены в 100 процентных ячейках, а информация про 78-96 остатки отсутствует полностью. Видимо, этот участок вообще не был включен в рассматриваемую структуру. В чем же дело? Скорее всего – дело в электронной плотности. Полученная модель, наверное, хорошо подходила для остальных аминокислот, а участок в 78-96 остаток просто вырезался из нее. Что было нехорошо. И, возможно, авторы сжульничали и просто выкинули этот кусок. Чтобы было лучшее соответствие. Но это только мое мнение. Я решил восстановить электронную плотность для цепочки C и посмотреть, как эти остатки вписываются в нее. Нет ли там возможного участка для пропущенной вставки. Однако, нет. На рисунке ниже слева вы видите электронную плотность для белкового остова, а справа – и для остова, и для боковых групп.
_3.png)
_4.png)
Так что, скорее всего, на самом деле электронной плотность для тех остатков 78-96 просто отсутствует, и видимо эти остатки выглядели как некая вставка. Поэтому, авторы их не включили в рассматриваемую модель.
Другое объяснение (и, наверное, более правдоподобное) состоит в следующем. Occupancy атомов аминокислот в соответствующем отрезке просто 0, по каким-то причинам, авторам не удалось нормально их померить. Ну а поскольку в таком случае не совсем понятно, как будет расположен этот отрезок, авторы посчитали нужным его просто выкинуть. Правда, я не уверен, насколько реальна подобная ситуация, когда occupancy атомов аминокислот из целого отрезка равны 0?
№3 – 2B5A
_1.png)
Год расшифровки: статья - Crystal structure of the restriction-modification system control element C.Bcll and mapping of its binding site. Год 2005. Журнал Structure.
Разрешение: 1.54 ангстрем (хорошее).
Странности: В этой структуре хорошее разрешение. Разрешение самого плохого рефлекса – 19.74 ангстрем. Полнота 99.5%. Это очень хорошо. R-value и R-free равны 0.165 и 0.201 соответственно. Пока все неплохо. Но есть и проблемы… Если посмотреть на 21 глутамин цепи C, то становится непонятно, что это…
Такое ощущение, что две боковые группы глутамина растут из одного С-alpha атома. Посмотрите следующую картинку, чтобы в этом убедиться.
_2.png)
Как так вышло? Давайте посмотрим на соответствующую запись PDB для этого остатка.
_3.png)
Как видно, тут лежат сразу две альтернативные конформации аминокислоты глутамина. Почему? Ответ дает коэффициент заселенности (он обведен красным) – для нескольких атомов он равен 50 процентов. То есть в половине ячеек глутамин в таком положении, в половине – в другом. И авторы не смогли решить, какой из них правильный, потому положили оба. А вот как выглядит соответствующая электронная плотность для данного неопределившегося остатка.
_4.png)
1 структура – 3WDN (PDB ID). Определена в 2014 году. Разрешение – 0.86 ангстрем. R-factor – 0.114, а R-free – 0.139.
2 структура – 3WUB (PDB ID). Определена в 2014 году. Разрешение – 2.08 ангстрем. R-factor – 0.196, а R-free – 0.221.
Для сравнения двух структур по числу атомов с плохой заселенностью и с плохим температурным фактором мною был написан скрипт, который принимает на вход два pdb файла и выдает количество соответствующих атомов и общее число атомов в структуре.
Для двух структур, представленных выше, скрипт выдал следующие данные:
Number of atoms with bad occupancy (<1) in 3WDN is 399
Number of atoms with bad occupancy (<1) in 3WUB is 0
Number of atoms with bad temp factor (<30) in 3WDN is 28
Number of atoms with bad temp factor (<30) in 3WUB is 1467
Number of atoms in 3WDN is 1172
Number of atoms in 3WUB is 2442
Результат ожидаемый. В структуре с хорошим разрешением мы видим, что есть приличное количество атомов с плохим значением заселенности. В то время как у структуры с плохим разрешением число таких атомов равно 0. Дело тут в том, что при хорошем разрешении мы увидим два заметных пика, отвечающих двум возможным положениям этих атомов (эти атомы представлены в разных ячейках в разных положениях). И в файле структуры с хорошим разрешением это можно отметить, поставив соответствующее число occupancy. А температурные факторы у такой структуры в основном хорошие, всего лишь 28 атомов имеют плохой температурный фактор.
В структуре с плохим разрешением все атомы имеют occupancy 1. Авторы не видят отдельных пиков. Но если в структуре все-таки есть такие атомы, которые в разных ячейках представлены в разных положениях, то пик соответствующего атома будет просто размыт (на плохом разрешении). И авторы отмечают такие атомы высоким температурным фактором. У структуры с плохим разрешением число таких атомов достигает больше половины от всех атомов!