AE015929 STAES 1598006-1599559 (compl.)
BA000018 STAA1 506161-507715
AL591974 LISMO 37466-39020
BA000043 GEOKA 30790-32343
AE016879 BACAN 9335-10841
AL009126 BACSU 9810-11364
AE004092 STRP1 17170-18504
FM211187 STRPN 16806-18218
1. Для объединения всех последовательностей в единый файл воспользовался:
cat *.fasta > all.fasta
2. Провел выравнивание программой muscle:
muscle -in all.fasta -out all_aligned.fasta
3. Далее закинул выравнивание в MEGA и построил дерево с помощью Neighbor-Joining.
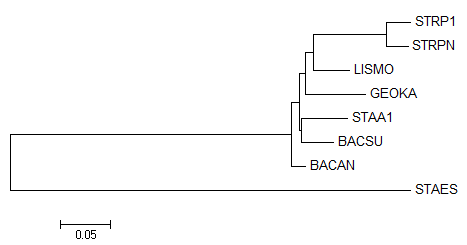
Дерево не совпало с правильным. Более того, оно содержит очевидные ошибки. И получается, что результат намного хуже тех, что были получены с белковыми последовательностями. Этому можно найти объяснение. Ведь качество нуклеотидного выравнивания по сравнению с белковым недостаточно высокое. Не существует никаких функциональных групп нуклеотидов (как у аминокислот). Замена одного нуклеотида на любой другой влечёт за собой одинаковый штраф. И видимо, в этом подходе нельзя сказать с уверенностью, является ли определённая нуклеотидная замена важной для разделения соответствующих видов в эволюционном плане. Это ведёт к соответствующим погрешностям.
Одна ветка {STAES} против {BACAN, BACSU, STAA1, GEOKA, LISMO, STRPN, STP1} уже бросается в глаза. Она неверная.
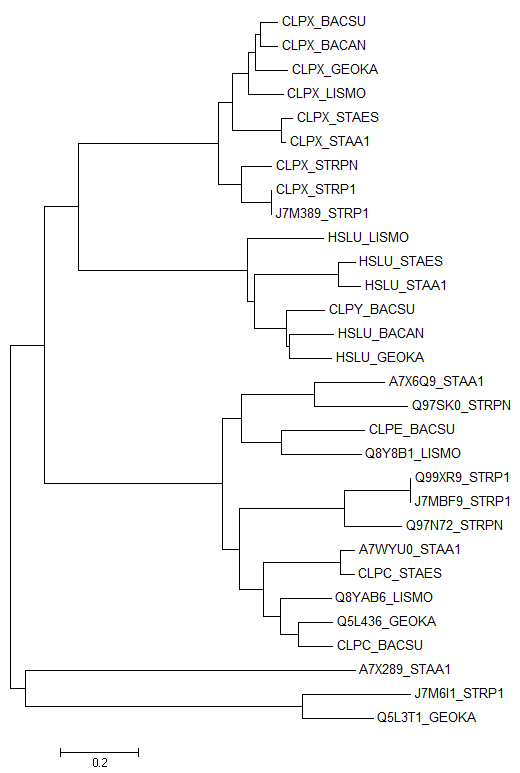
1. Для поиска гомологов воспьлозавлся программой blastp:
blastp -query clpx_bacsu.fasta -db prot -out gomolog.fasta -evalue 0.001
2. Получил последовательности благодаря сервису retrieve на сайте UniProt и выровнил их программой muscle
3. Построил дерево, используя программу MEGA методом Neighbor-Joining. Нашлось 30 гомологов.