STRIDE
Определение вторичной структуры выбранного мною белка 1MKI производилось с помощью программы STRIDE. Предлагаю ознакомиться с визуальной выдачей этой программы:
Также привожу ссылку на выдачу программы в текстовом виде.
Описание альфа-спирали
Давайте опишем первую альфа-спираль, которую выдает нам STRIDE для цепи А, причем, я имею в виду именно альфа-спираль, а не другие варианты (как, например, спираль 310, которую выделил STRIDE, на картинке вверху обозначена синим цветом). Программа выдала, что эта альфа-спираль определена следующими границами: 13 и 25 аминокислота. А вот спираль с 26 по 31 остаток – это спираль 310. Давайте теперь посмотрим на то, что лежит в PDB файле! Там для этой же спирали приведены границы 13 – 26 аминокислота (указано, что это просто helix, без уточнений альфа или 310), и еще указано, что есть спираль с границами 27 – 32 аминокислота (также указана, как просто helix). То есть, границы сдвинуты на одну аминокислоту!
Можно в PyMol посмотреть на то, как выглядит данная спираль! Она представлена на картинке внизу:
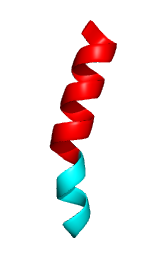
Красным выделена альфа-спираль с 13 по 25 остаток (по STRIDE), циановым – спираль с 26 по 31 остаток. Виден характерный загиб, в принципе, можно даже на взгляд определить, где кончается нормальная альфа-спираль и начинается спираль 310. Последний тип спиралей в белках не так уж редок, STRIDE в одной цепи белка 1MKI насчитал 6 штук. Правда, они, как правило, короткие (и деформированные из-за стерических напряжений).
Бета-лист
Если мы посмотрим на бета-лист так в цепи А так, как его представляет pdb файл, то увидим нечто вроде такой картины:
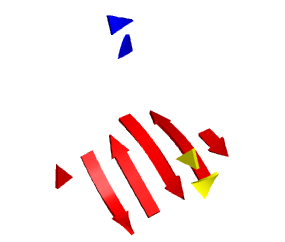
В pdb файле указаны 10 бета-тяжей, образующих 3 листа (6 в одном – красный – лист А, по 2 в оставшихся – синий (лист B) и желтый (лист C)). На самом деле, 5 из этих бета тяжей состоят из 2 остатков (включая синий и желтый листы на рисунке, там все тяжи из двух остатков), так что не совсем понятно, правомерно ли это считать полноценными бета-тяжами.
Теперь посмотрим, какие бета-листы выделил STRIDE. Самым наглядным образом это можно сделать при сравнении тех бета-листов, что лежат в PDB. На картинке внизу это и изображено (красный – бета-листы из PDB файла, синий – бета-листы из STRIDE).
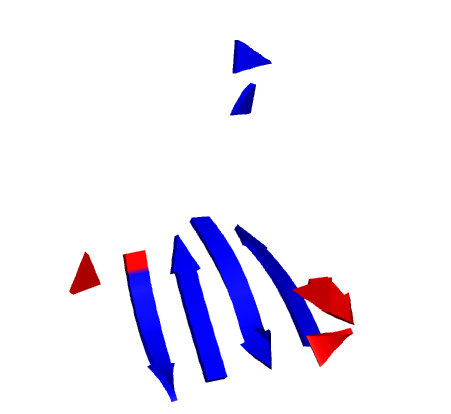
Видно, что тут совпадают только 2 бета-листа (А и В). И то не полностью. Самый крупный бета-лист представлен и в PDB, и в STRIDE, однако в последнем в нем содержится меньше бета-тяжей.
Бета-мосты
STRIDE выделил на последовательности 4 остатка, которые он отнес к бета-мосту. Привожу их изображение (они выделены оранжевым) и водородные связи между ними.
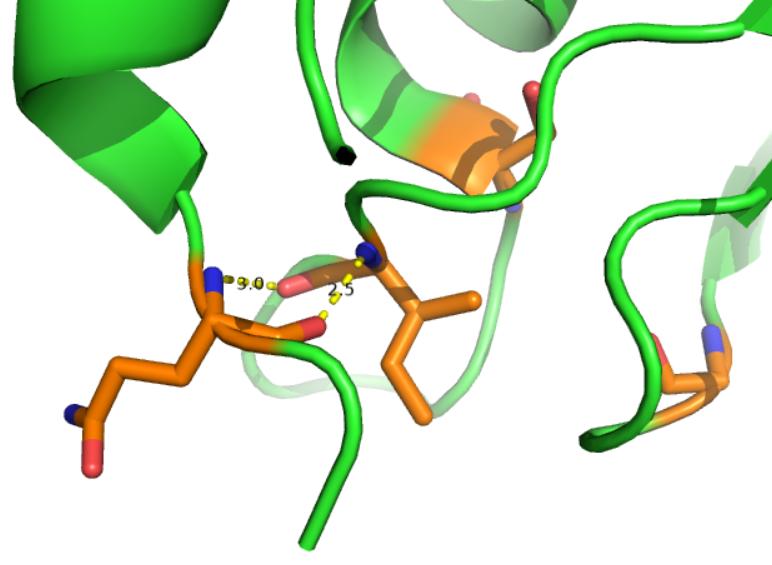
Здесь не совсем понятная ситуация, так как в определении бета-моста говорится, что это простая форма водородной связи на подобии той, что есть в бета-листах, объединяющая, как правило, 3 остатка (i-1, I, i+1 и j-1, j, j+1 – и если между ними есть пара водородных связей) [http://www.bioinfo.de/isb/2007/07/0037/main.html]. Приведенная же выше картина не совсем похожа на описанную.
SHEEP
Теперь, воспользовавшись программой Sheep, я рассмотрел лист А из моей структуры 1MKI. Привожу карту этого листа:

А вот изображение, которое показывает этот же лист в Jmol:
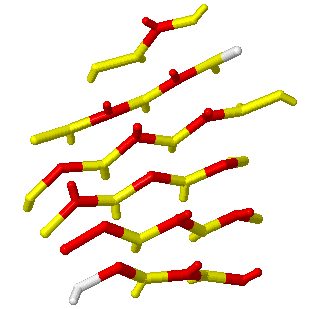
Цвета здесь соответствуют тем, что приведены в схеме бета-листа. В принципе, тут и так все наглядно, какая колонка, какому хребту соответствует. Но чтобы было совсем понятно, я приведу следующие изображения:
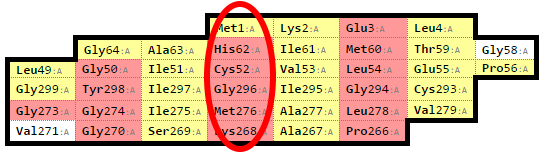
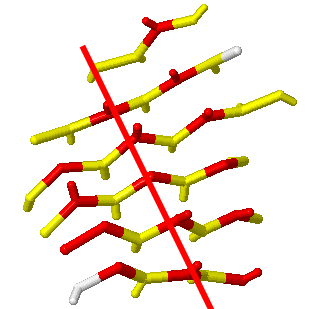
Колонка в схеме, для которой показано соответствие в Jmol, выделена красным овалом. А хребет в Jmol для данной колонки выделен красной линией.
Изображения внизу демонстрируют расположение листа А цепочки А (выделена красно желтыми цветами) в глобуле белка.
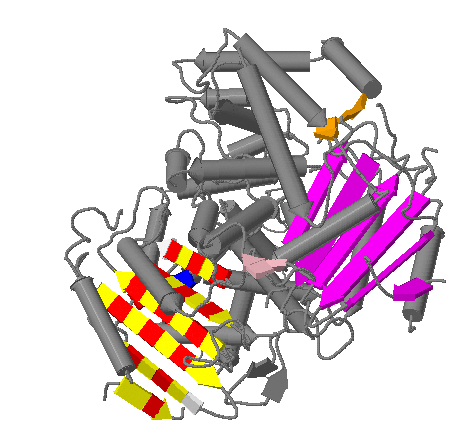
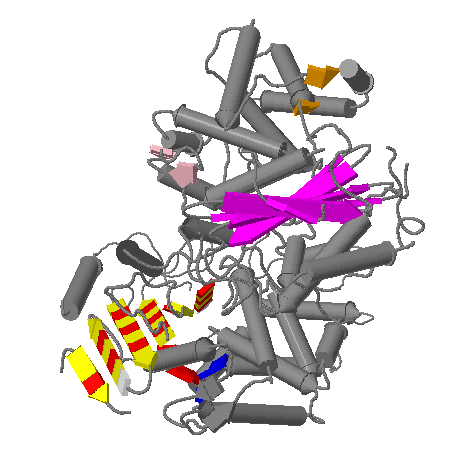
Я привожу эти картинки чтобы показать, что почти весь бета-лист лежит внутри глобулы, что и логично. Боковые группы остатков, помеченных желтым – направлены вверх, а красных – вниз. Убедиться в этом можно на изображении, приведенном ниже:
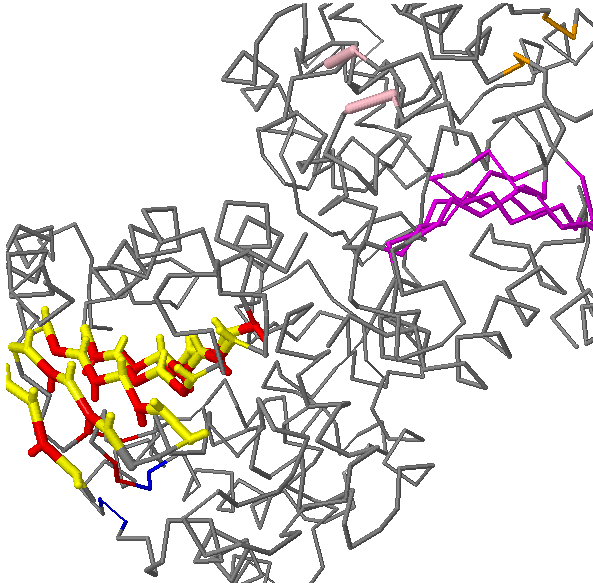
В связи с этим я не знаю, как ответить на вопрос, по поводу направленности боковых групп. С одной стороны, для почти всех бета-тяжей здесь, остатки направлены к гидрофобному ядру. Но с другой, можно также грубо предположить, что именно красная сторона смотрит в глубь белка, а желтая (все-таки) на поверхность (в задании d3 будет показано, что и те, и другие боковые группы смотрят на гидрофобные кластеры).
И в заключении, если судить о том, как определил Sheep границы бета-листа А, по сравнению со STRIDE: Sheep выделил здесь 6 тяжей (а STRIDE – 4), также, как в PDB файле, только с более расширенными границами.

Рисунок выше показывает, как не совпадают границы бета-тяжей, лежащих в PDB, и тех, что определил Sheep. А несовпадение данных PDB и результатов STRIDE приведены выше.
Cовмещение структур моего белка (1MKI) и четырех структурных гомологов
На сервере PDBeFold я выбрал pairwise 3D alignment. В качестве query я выбрал свою структуру – 1MKI, а в качестве target – whole pdb archive. Структурных гомологов было решено искать только для цепочки А. Lowest acceptable match был выбран по умолчанию 70%.
Были получены следующие результаты:
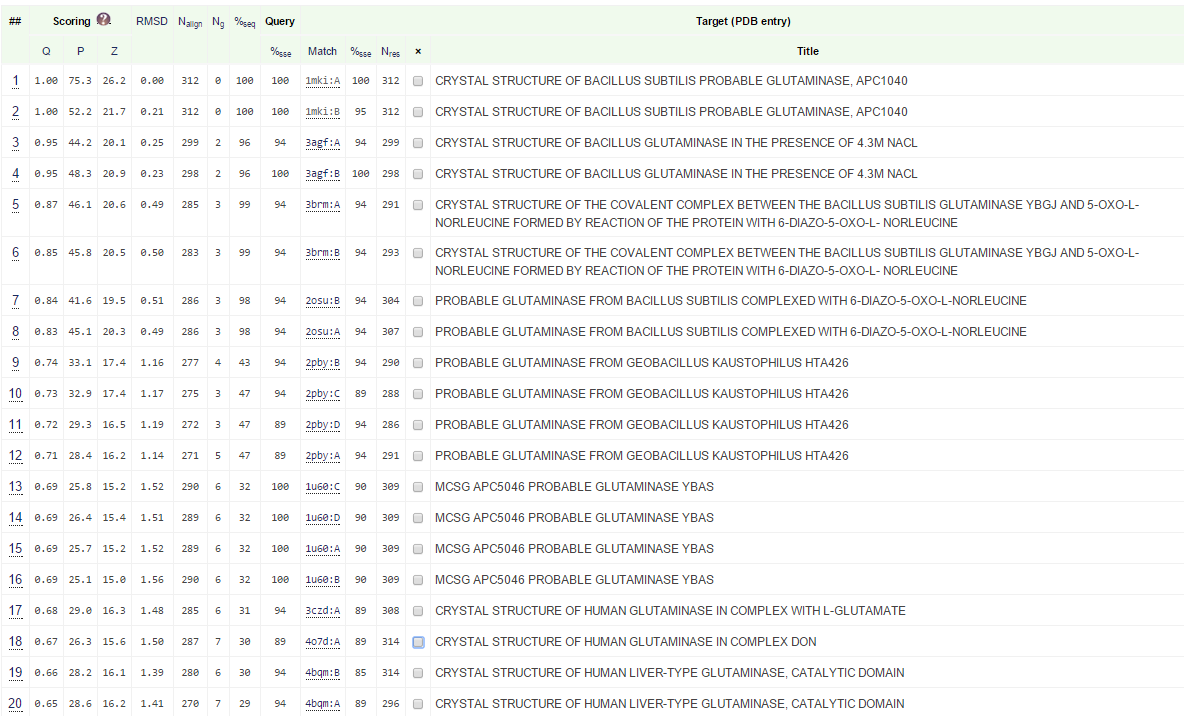
Далее, была предпринята попытка выбрать 4 структуры для множественного выравнивания с (1MKI). Это 2PBY цепь А (seq identity (si) 47%, secondary structure identity (ssi) 89%), 1U60 цепь А (si 32%, ssi 100% - обратите внимание, какое малое сходство по последовательности, однако 100 процентное сходство по структуре) 3CZD цепь А (si 31%, ssi 94%) и 3SS3 цепь B (si 30%, ssi 89%). Все эти молекулы являются глутаминазами (как и входная структура), бактериальные и эукариотические.
Представляю изображение совмещения всех 5 структур сразу:
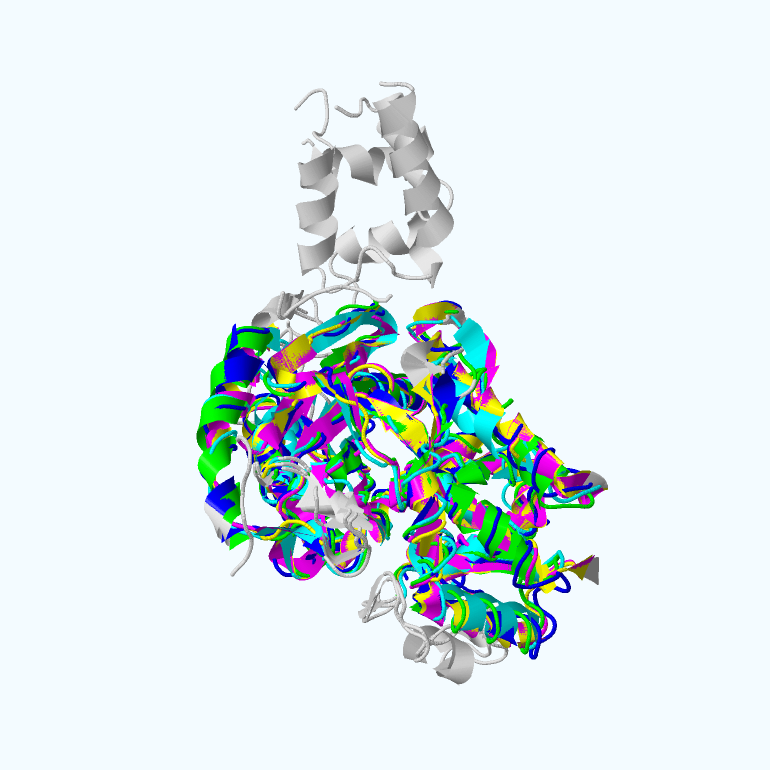
Как видно, в принципе все структуры очень схожи, за исключением некоторой части, которая окрашена в серый цвет.
Теперь сравнение выравниваний по структуре и по последовательности (с помощью Muscle). Во-первых, сразу надо отметить, что в структуре 1MKI отсутствуют 2 участка (с 6 по 12 и с 36 по 44 остатки), поэтому, это накладывает свое отличие на выравнивания. Причем, это касается не только 1MKI, но и, например, 2PBY (отсутствуют остатки с 93 по 108). В связи с этим различий получается довольно много. Внизу представлен пример того, как эти различия влияют на выравнивания.
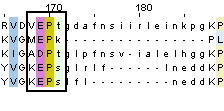
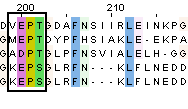
Слева – выравнивание по структурам, справа – выравнивание по последовательностям. Прямоугольником выделен одинаковый участок в обоих выравниваниях (выровнены одни и те же остатки!). Но из-за того, что в 2PBY пропущен достаточно большой фрагмент, в выравнивании по структурам получается солидный промежуток из гэпов.
Если закрыть глаза на это, то все остальное выровнено практически одинаково, за исключением некоторых мелочей. Например, вот такой случай.
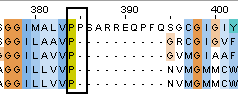
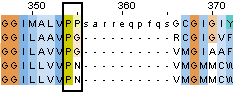
Слева – структурное выравнивание, справа – по последовательности. Показан один и тот же участок, по другим колонкам видно совпадение. Но есть небольшое отличие, и оно показано в черном прямоугольнике. В нем лежит 2 колонки для выравнивания структурного, и только одна в выравнивании по последовательностям. Но сама колонка, на самом деле, есть и в том, и в другом выравнивании. Просто в случае структур в этой колонке есть пролин, а в случае последовательностей – пролин заменен на серин. Честно говоря, даже не знаю, что здесь лучше – пролин или серин для структурного выравнивания, программа решила, что пролин! И, наверное, так и есть. Привожу картинку совмещения пролина 1mki с глицином 2bpy:

Зеленый остов – 1MKI (синим показан остаток пролина 281), желтый – 2PBY (розовым – остаток глицин).
СОВМЕЩЕНИЕ ПО ЗАДАННОМУ ВЫРАВНИВАНИЮ
Для этой работы я выбрал структуру 1OGA. Согласно SCOP в альфа-цепи (которая в данной структуре обозначается как D) границы константного домена лежат с 118 по 202 остатки. А в бета-цепи (которая в данной структуре цепь Е) - с 119 по 245 остатки. Интересно, что Pfam определяет домены следующим образом: в альфа цепи лежит домен DUF1968 (PF09291) – домен с неопределенной функцией на границах 118-201 (почти аналогично SCOP); в бета цепи лежит домен С1-set (PF07654) – границы 129-222 (немного отличается от SCOP). Последний домен – это непосредственно и есть константный домен иммуноглобулина!
Привожу изображение из Jmol Pfam:
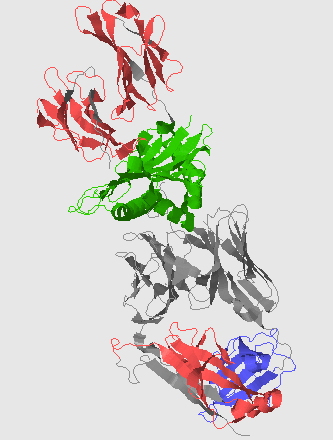
На самом деле, здесь представлена вся структура 1OGA. Интересующие нас цепи расположены в самом низу! Там домен альфа-цепи окрашен в синий цвет, а домен бета-цепи – в красный.
Привожу изображение только двух выбранных доменов, построенное в PyMol:
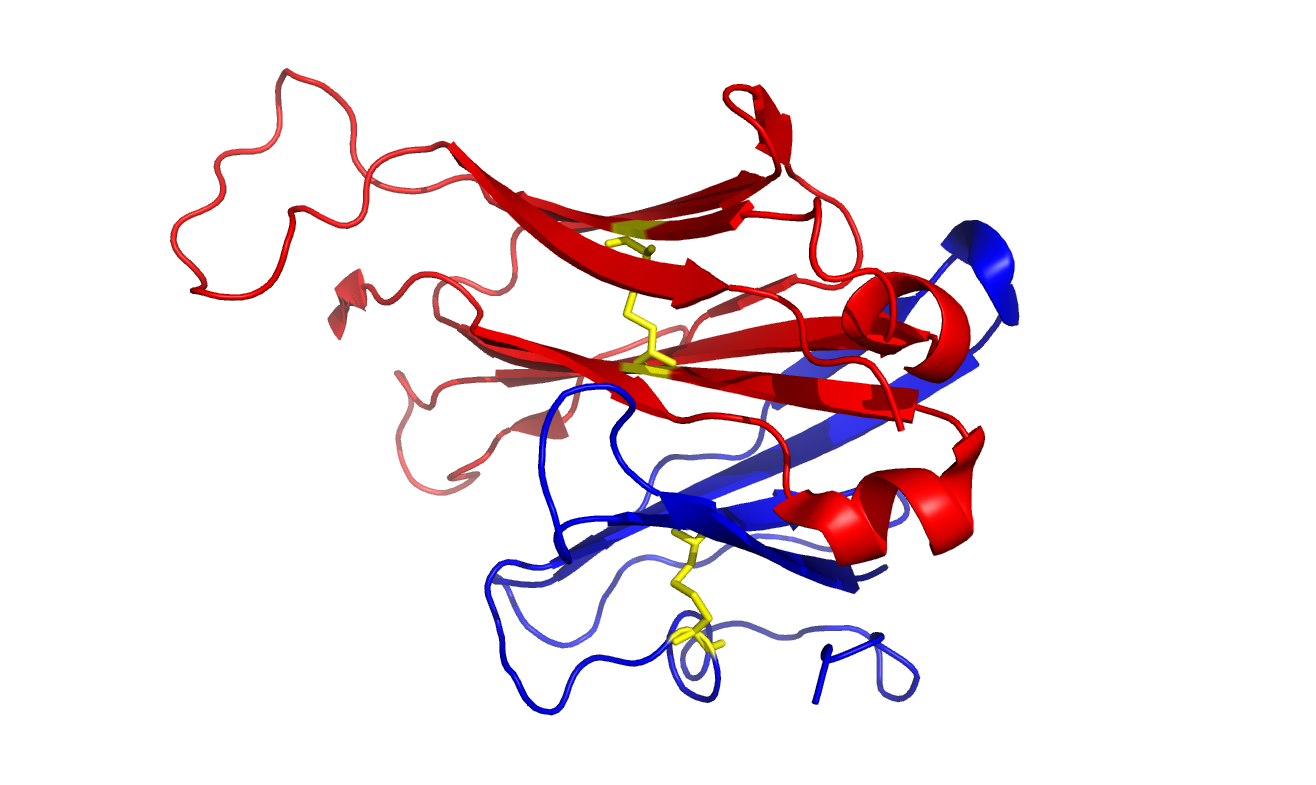
Домены окрашены в те же цвета, что и на изображении выше (синий – домен альфа цепи, красный – домен бета цепи). Желтым показаны остатки цистеина, образующие дисульфидный мостик. Видно что одном домене только один бета-лист, а в другом – два бета-листа. И предстоит выяснить, какие бета-листы из разных доменов соответствуют друг другу. И для этой цели используем программу Sheep для соответствующих доменов (для этого были созданы отдельные файлы pdb, в которых хранились эти структуры). Но на самом деле, если судить по изображениям выше, в этих бета-листах мало общего…
Sheep, как и ожидалось, выделил всего один бета-лист для домена из альфа-цепи. В ней есть так-называемый beta-buldge в виде лизина 158 (и аспарагина 157?), который выглядит, как вставка. На мой взгляд, он тут лишний, программе его выделять не следовало, он никак картину не меняет).
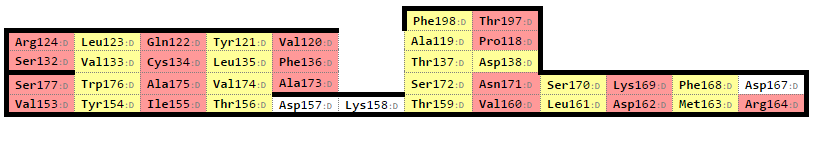
А вот для бета-цепи (тоже, как и ожидалось) удалось выявить два бета-листа, которые приведены ниже.
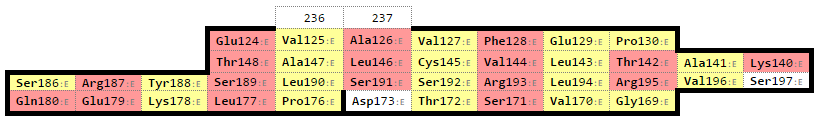
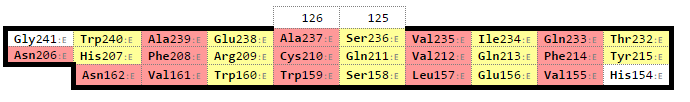
И что? Кажется, что между этими листами нет ничего общего! Единственное, что их объединяет – это атом цистеина в середине центрального бета-тяжа. Я попробую каждый лист из бета-домена с листом из альфа, посмотреть, где какое будет наложение.
Давайте посмотрим более внимательно на лист из домена альфа-цепочки! Привожу его изображение из PyMol.
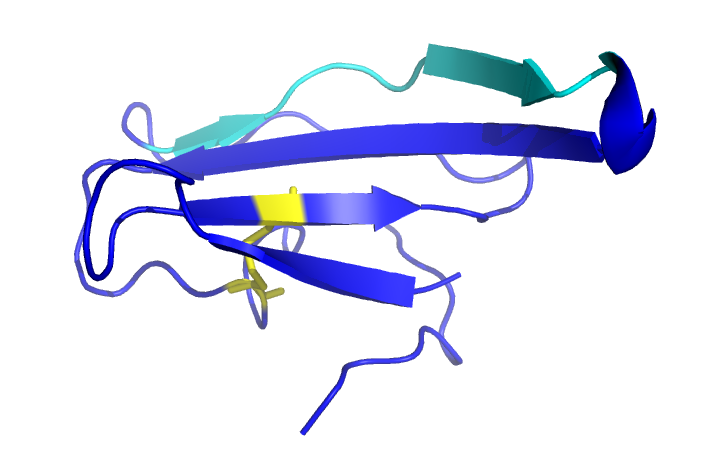
Циановым выделены остатки, которые занимают последнюю строчку на соответствующей карте (см. выше). Видны те самые нерегулярности, из-за которых на карте получился гэп. На мой взгляд, в лист здесь входят только три тяжа, полноценные три тяжа.
Поэтому для начала я постараюсь выровнять этот бета-лист со вторым бета-листом из домена бета-цепи (в котором 3 тяжа). Но для начала надо добиться, чтобы они были в одинаковой ориентации.
Для домена альфа цепи:
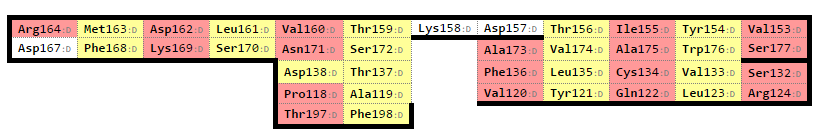
Для второго бета-листа из бета цепи:
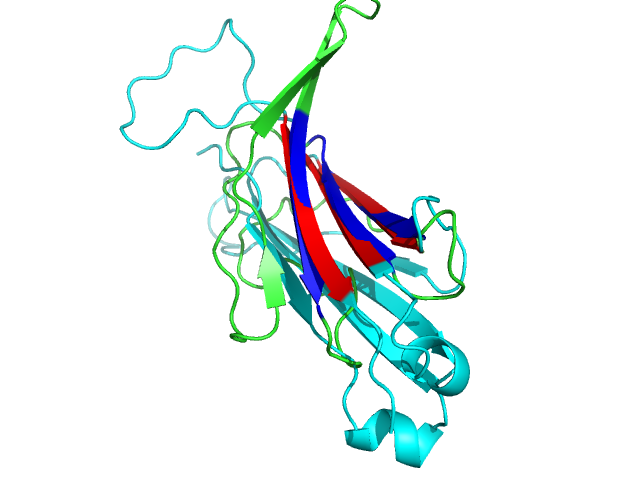
Обращаю ваше внимание, что для листа из альфа-спирали я рассматриваю только центральные три тяжа!!! Самый верхний и самый нижний я не рассматриваю. И в соответствующем бета-листе из бета цепи тоже только 3 тяжа – их ориентация совпадает с ориентацией центральных бета тяжей в листе из альфа цепи.
Консервативные остатки цистеина здесь, образующие дисульфидные связи – это (для домена альфа цепи) цистеин 134, и (для листа из домена бета цепи) цистеин 210.
Теперь попробуем построить структурные выравнивания этих двух бета листов. При выборке атомов, которые должны быть выровнены друг с другом, я ориентировался на консервативные цистеины, спаренные с ним остатки и их соседи. В итоге я использовал следующие команды:
select ch1, 1oga-a and (resi 118-124 + resi 132-138 + resi 171-177) and name ca (альфа цепь)
select ch2, 1oga-b and (resi 155-161 + resi 208-214 + resi 233-239) and name ca (бета цепь)
pair_fit ch1, ch2
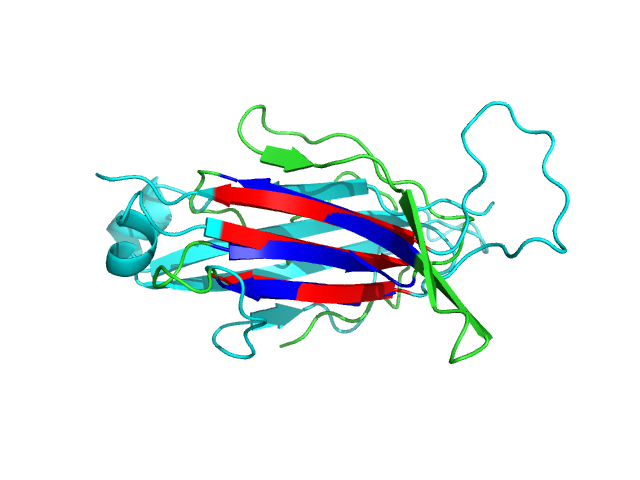
Изображения выше показывают полученный результат наложения. RMSD = 1.158. Красный – атомы для листа из бета-цепи, а синий - для листа из альфа-цепи. Номера остатков прописаны в командах (соответствующие номера) и они брались из карты.
Ниже приведены ссылки для скачивания соответствующих pdb файлов и скрипт, который строит структурное выравнивание:
Теперь, что касается второго листа из домена в бета-цепи…
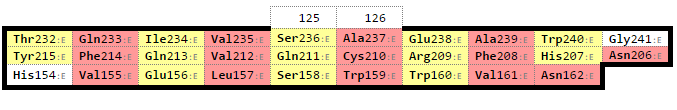
Привожу карты бета-листов в одинаковой ориентации. Тут уже рассматривались 4 тяжа (в листе для альфа цепи не рассматривался тяж из 2 остатков).
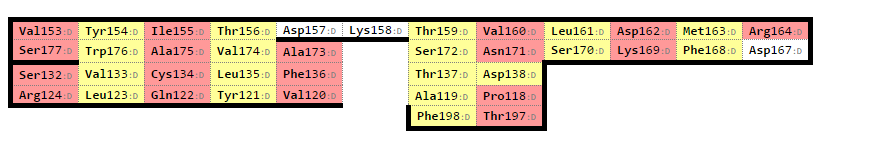
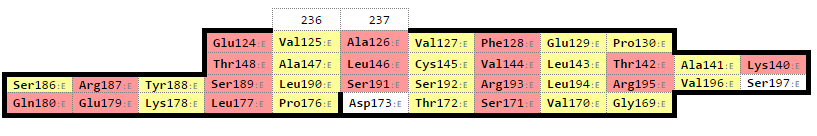
Консервативные остатки цистеина - 134 (для альфа цепи) и 145 (для бета цепи).
Привожу команды, в которых также отображены номера остатков, используемых для выравнивания.
select ch1, 1oga-a and (resi 118-123 + resi 132-137 + resi 171-176 + resi 153-158) and name ca (альфа цепь)
select ch2, 1oga-b and (resi 169-173,176 + resi 190-195 + resi 142-147 + resi 124-129) and name ca (бета цепь)
pair_fit ch1 ch2
Привожу результат:
Здесь уже выравнивание получилось так себе. RMSD = 2.399, хуже чем в предыдущем случае. Правда, что удивительно, в данном случае уже наблюдается сходство топологий – петли хоть и не выравниваются нормально (точнее почти не выравниваются), однако повторяют ход друг друга. Возможно, именно эти листы и соответствуют друг другу, несмотря на плохое наложение. скрипт
Обращаю внимание, что в скрипте используются те же файлы pdb, ссылка на которые представлена выше!
Нахождение гидрофобных кластеров производилось с помощью веб-сервера Clud. В качестве белка использовал 1MKI. При самом первом запуске оставил все параметры по умолчанию (порог расстояния и порог размера кластеров), чтобы посмотреть, что получится в этом случае (кластеры искались по всей структуре).
Демонстрирую изображение для самого крупного получившегося гидрофобного кластера (1160 атомов, в то время, как в самой структуре – 4863 атома).
Вот как представляет Jmol структуру 1MKI без гидрофобного кластера:
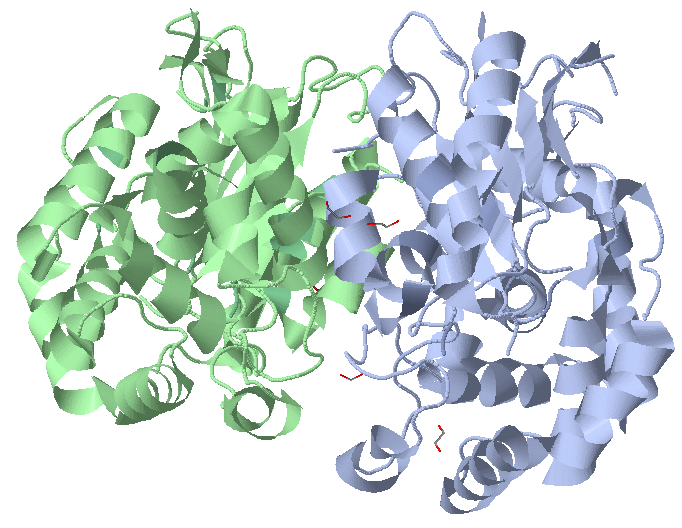
А вот в виде желтых шариков атомы, которые входят в определенный гидрофобный кластер:
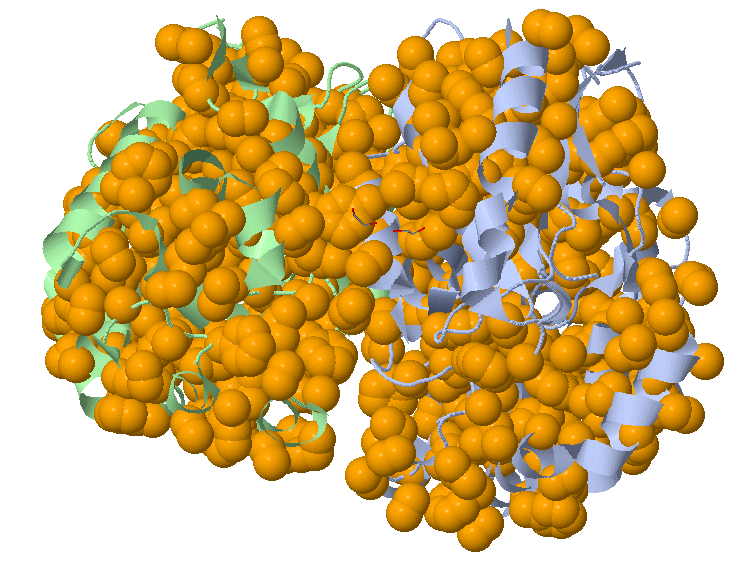
Та же картина, но в другом виде. Зеленый и синий – атомы цепей А и В, красные – атомы, входящие в гидрофобный кластер.

Обращает на себя внимание тот факт, что в получившемся результате достаточно много молекул из гидрофобного кластера лежат на поверхности:
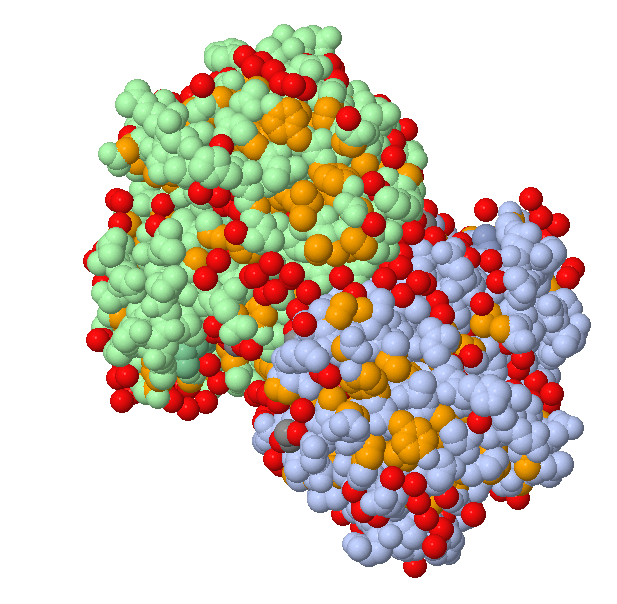
Здесь оранжевые шарики – те, что принадлежат гидрофобному кластеру. Видно, что достаточно приличное количество лежит на поверхности молекулы.
А вот второй по величине гидрофобный кластер имеет размер всего лишь 34 атома, что несравненно мало по сравнению с первым. Привожу его изображение (атомы кластера выделены синими шариками).

Теперь попробуем менять параметры! Самое интересное, что если поменять порог расстояния с 5.4 до 5, а минимальный размер кластера оставить прежним, то самый большой кластер будет иметь 801 атом – почти на 300 атомов меньше, чем у самого крупного в предыдущем случае.
Кластер из 801 остатка похож по форме на предыдущий самый крупный кластер, однако, в данном случае удалось найти 2 кластера по 135 и 130 атомов в разных цепях, которые не были найдены в прошлом случае. Привожу их изображение:
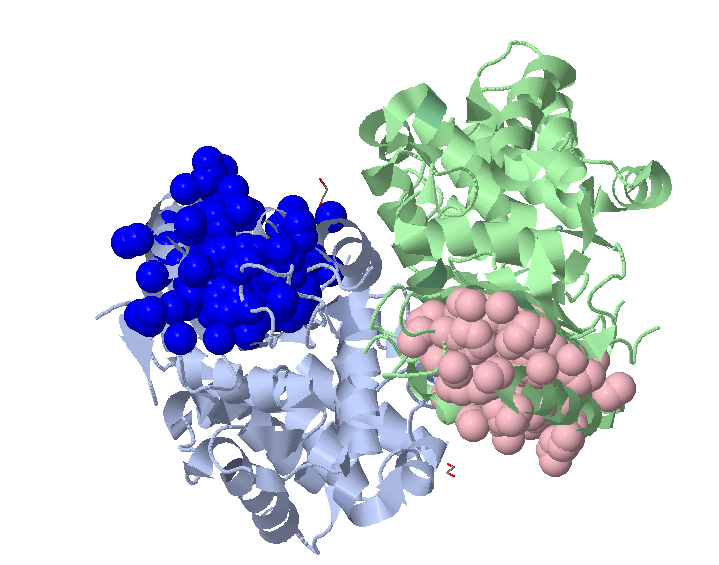
На самом деле, эти два домена симметричны, так как они в обеих цепях занимают одинаковое положение – между бета листом и альфа спиралью, которая лежит на границе белковой молекулы.
Если поставить порог на расстояние 4 ангстрема, то самый большой кластер будет состоять из 18 атомов:
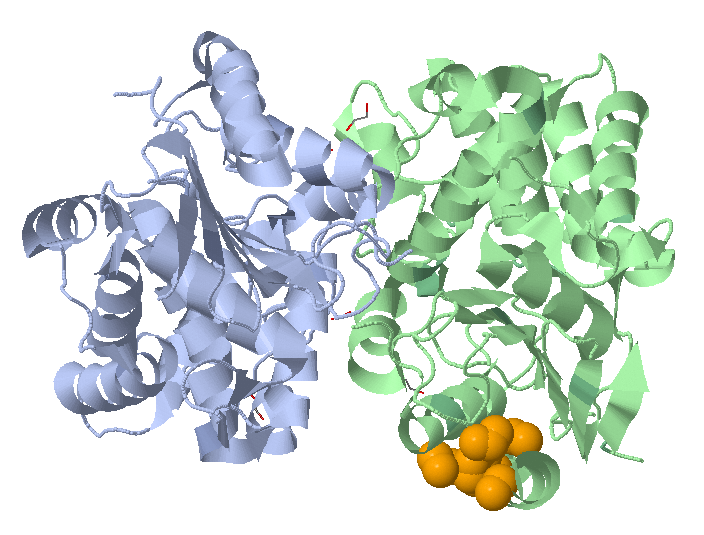
В данном случае кластер лежит между двумя альфа спиралями близко у поверхности.
Если поставить расстояние как 4.5 ангстрем, то самый крупный кластер будет иметь 417 атомов.
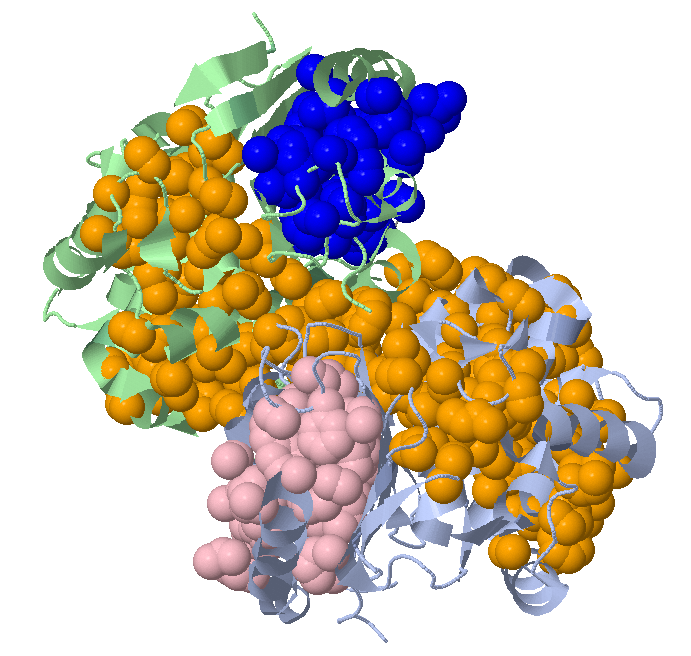
Самый крупный кластер здесь показан оранжевыми шариками, он симметричен, охватывает по сути две цепочки. Синие и розовые шарики – это те же самые 2 кластера, которые приведены выше. Правда тут у них не 135 и 130 атомов, а 95 в каждом.
И так, как видно из предыдущих картинок, самый крупный кластер охватывает почти всю молекулу белка (1160 атомов). А каждая цепочка молекулы в SCOP – это полноценный домен e.3.1.2 (Glutaminase). То же самое почти справедливо для домена из Pfam PF04960 (Glutaminase) (см. задание d5).
Попробуем теперь найти гидрофобные кластеры между двумя цепями в молекуле 1MKI.
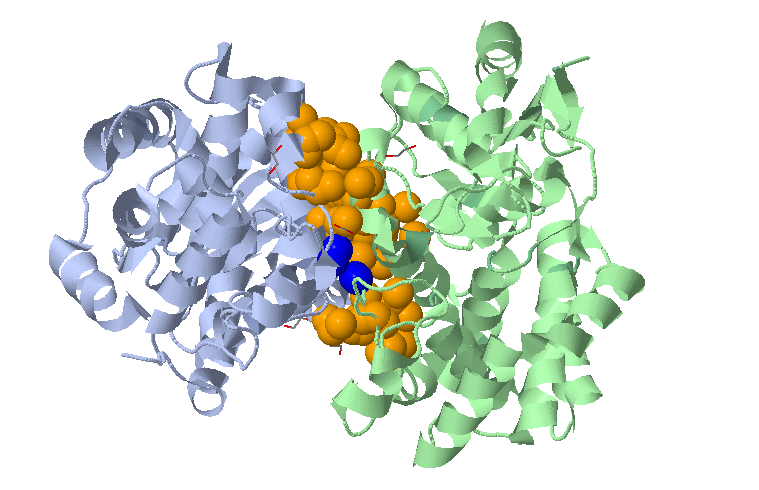
Программа нашла 2 кластера на границе (поиск производился с параметрами по умолчанию). Была найдено 2 кластера – самый большой (оранжевые шарики) – 76 атомов, самый маленький (синие шарики) – 4 атома. Видно, что 2 цепочки в молекуле держатся за счет гидрофобных взаимодействий.
Я работаю с ДНК-белковым комплексом, структура которого лежит в PDB с ID 1QPZ. Привожу структуру остова димерного белка в комплексе с молекулой ДНК:
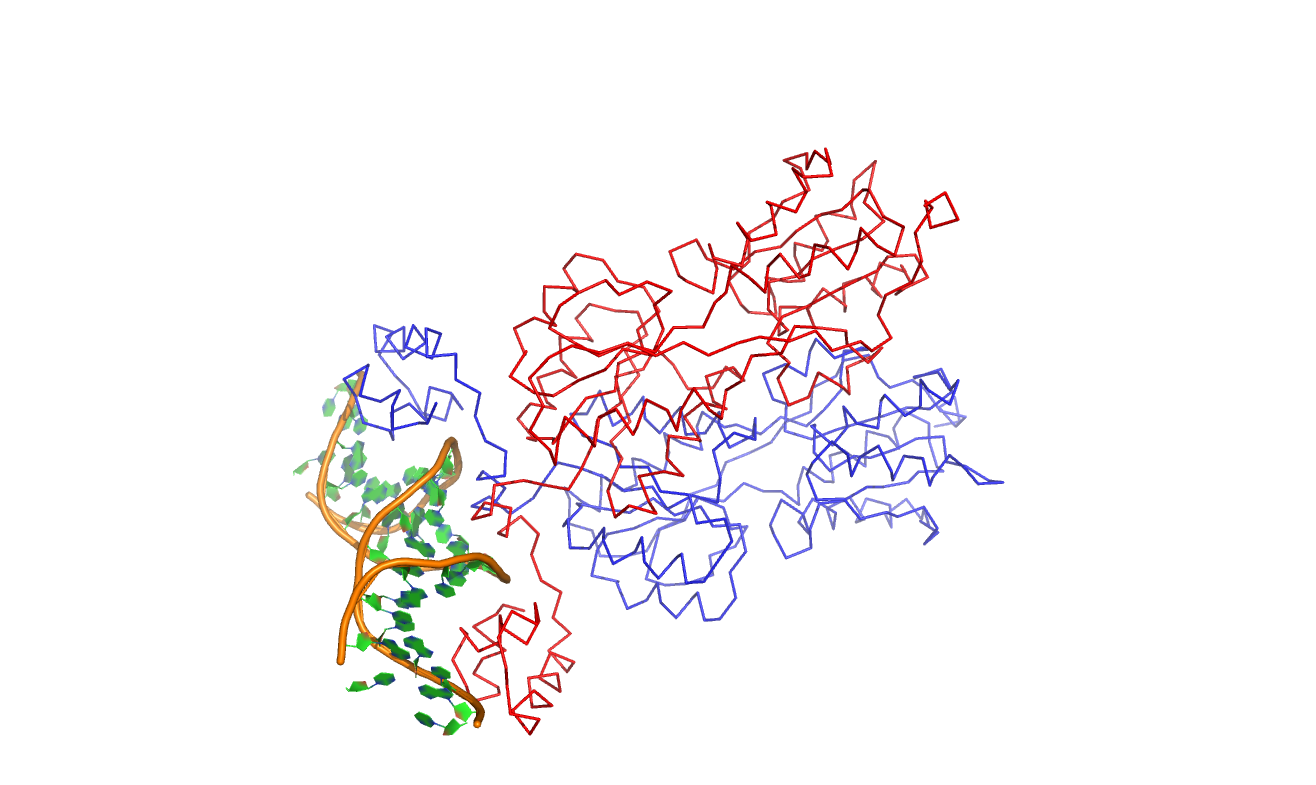
Попробуем создать поверхность контакта мономера белка с симметричным мономером на фоне остовной модели. Я не совсем понял, что тут имелось в виду, но привожу изображение контактирующих поверхностей двух мономеров на расстоянии 3.5 ангстрема:
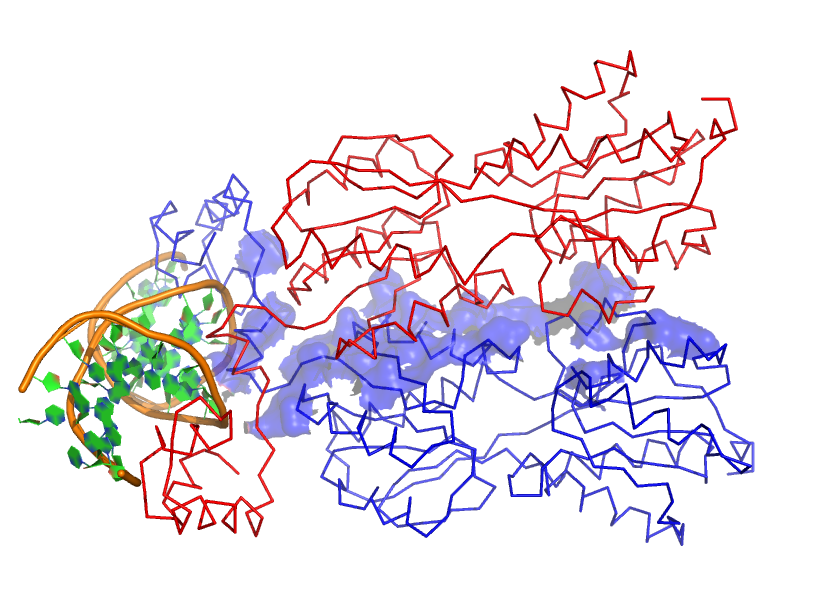
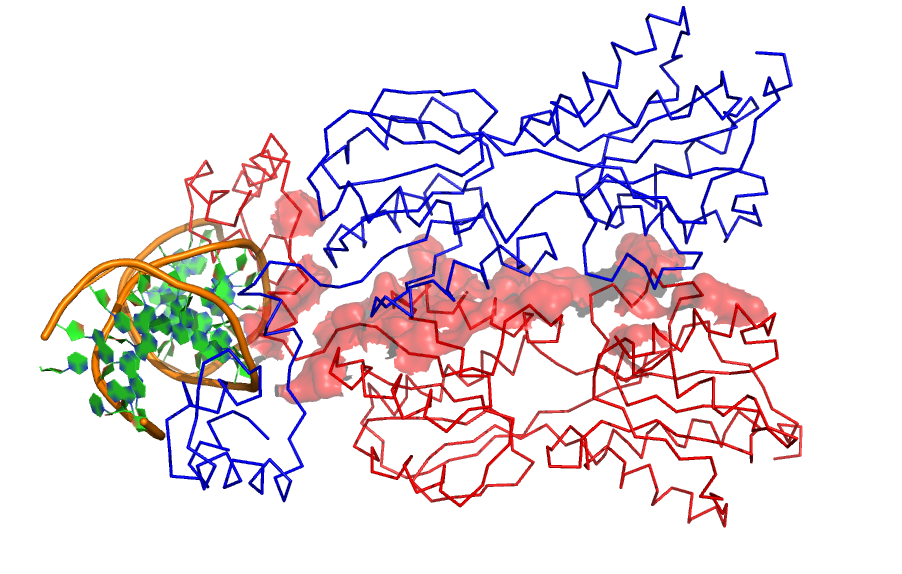
Синим цветом, слева, показана поверхность контакта одного мономера по отношению к другому (без ДНК!). Справа – аналогичный рисунок для другой субъединицы.
Теперь привожу изображения поверхности контакта димера с двойной спиралью ДНК на фоне остовной модели белка (слева), а также то же самое изображение на фоне проволочной модели двойной спирали (справа).
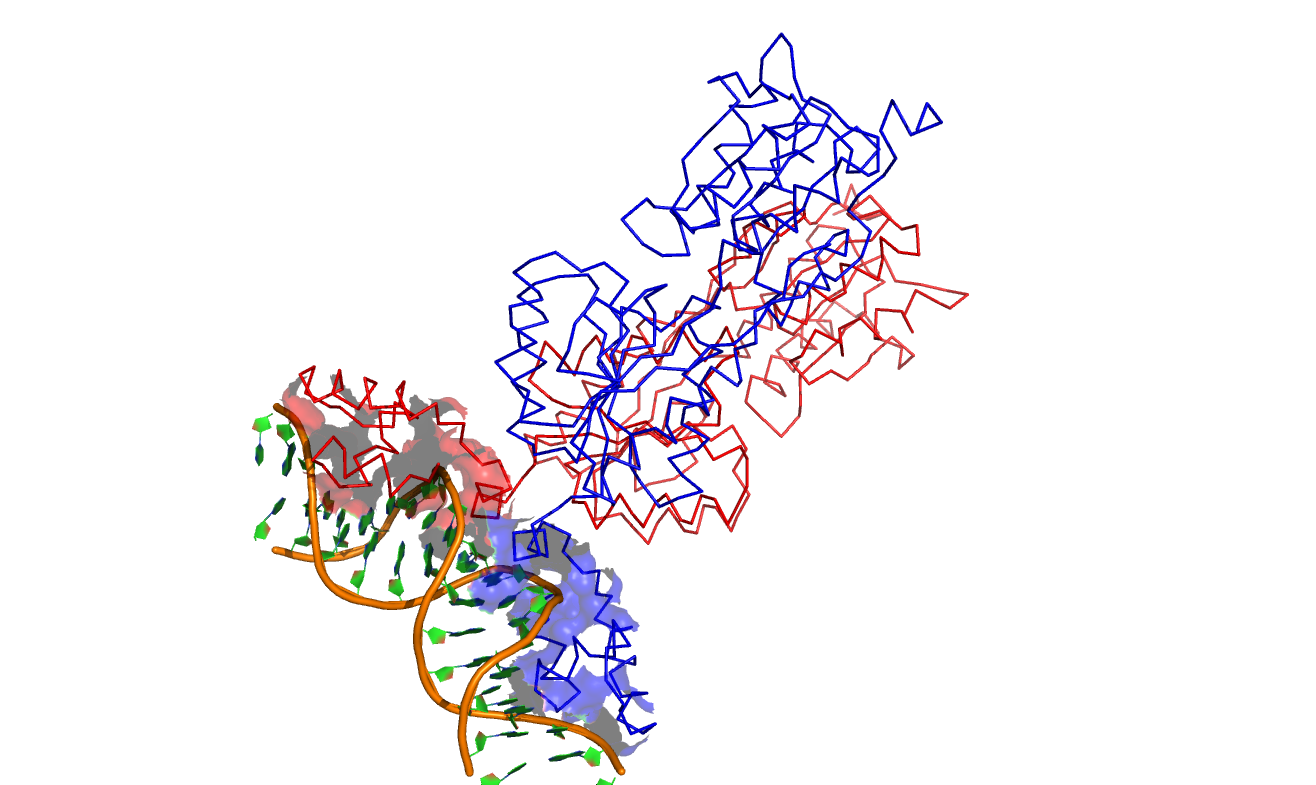
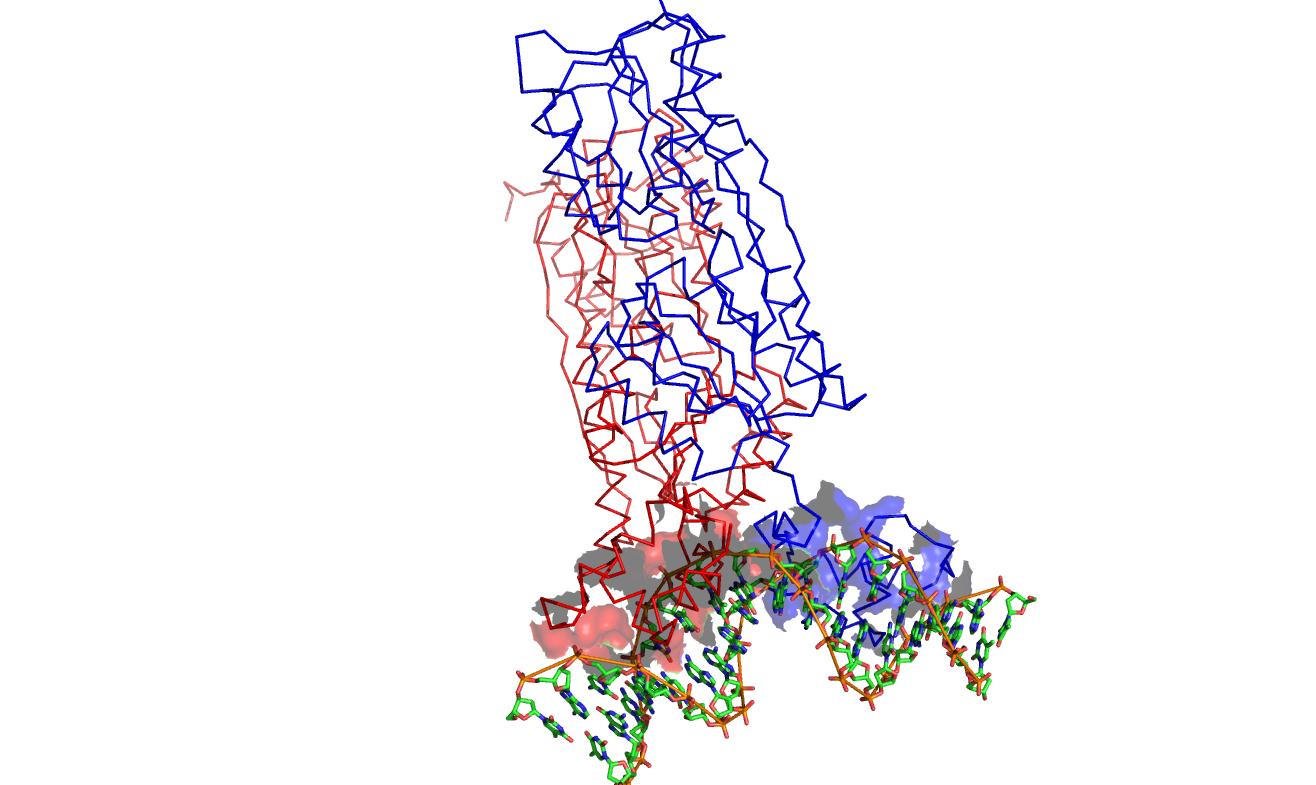
Синяя поверхность контакта – контакт ДНК и мономера, обозначенного синим цветом (аналогично с красной поверхностью).
Теперь давайте посмотрим, какой результат выдает программа Clud (поиск гидрофобных кластеров) для интерфейса двух мономеров в этом белке. Поиск производился с параметрами по умолчанию, кроме минимального количества атомов в кластере (было установлено в 10). На изображение ниже представлен результат Jmol:
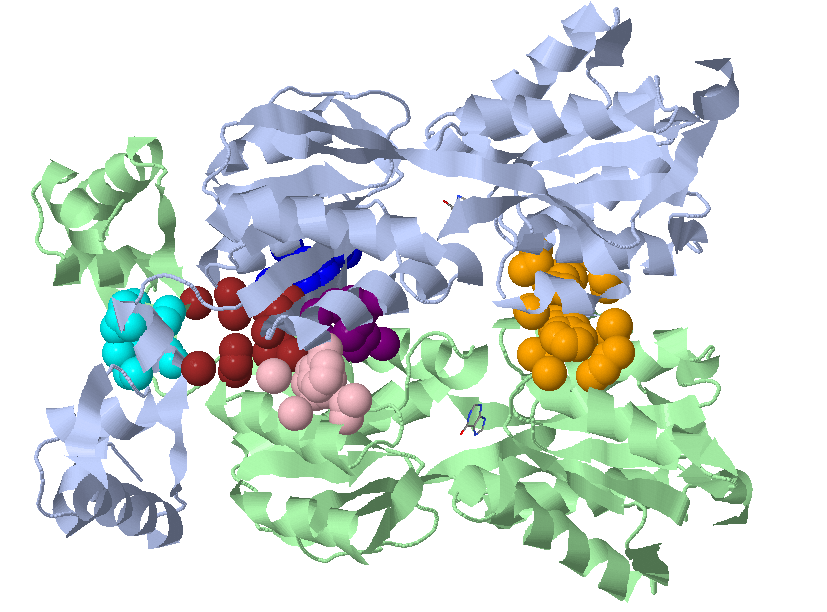
Самый большой кластер здесь выделен оранжевыми шариками, в нем 44 атома. Остальные кластеры поменьше: синий (18 атомов), бордовый (16 атомов), фиолетовый (16 атомов), розовый (18 атомов), циановый (14 атомов).
На рисунке ниже представлено изображение из Jmol – остовная модель, а также выделенная оранжевым поверхность контакта, в которую входят атомы из полученных гидрофобных кластеров.
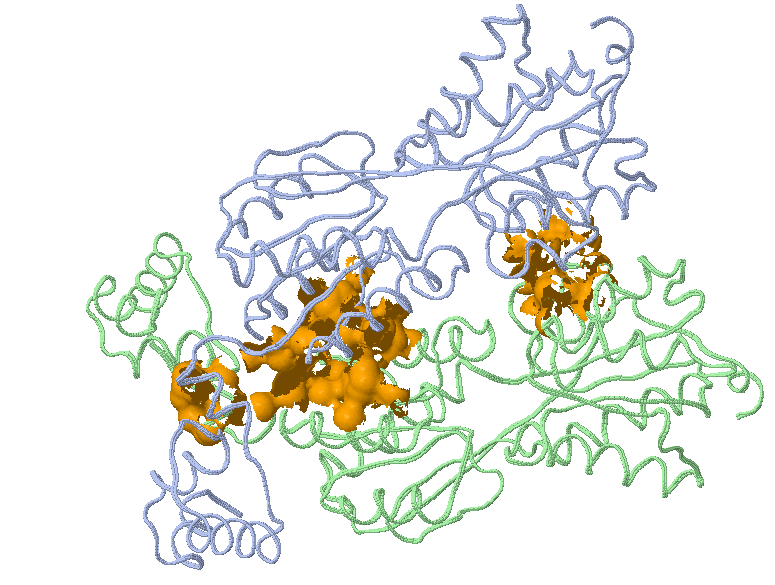
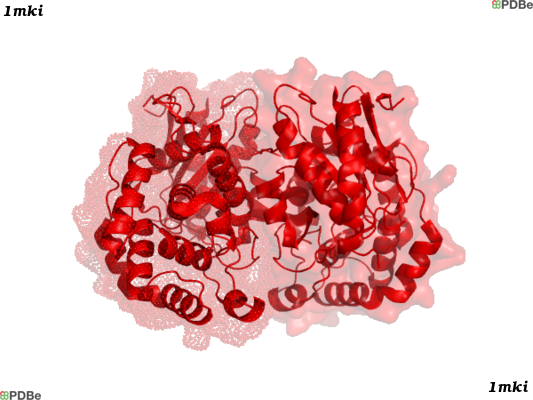
Я решил сравнить домены SCOP и Pfam для своей структуры 1MKI.
Этот домен называется e.3.1.2 (Glutaminase), и согласно SCOP, цепи А и В (полностью) моей структуры соответствуют этому домену, точнее - одна цепь - это целый домен. Ниже привожу изображение домена SCOP из PDBe (на самом деле, тут 2 домена – по одному на каждую цепь). Видно, что домен (окрашенный в красный) охватывает всю цепь.
Pfam тоже выделяет домен Glutaminase, как и Scop, но в отличие от последнего домен из Pfam не охватывает всю цепь целиком для данной структуры. Границы этого домена по Pfam с 32 остатка по 327. Ниже приведено изображение цепи А структуры 1MKI в PyMol.
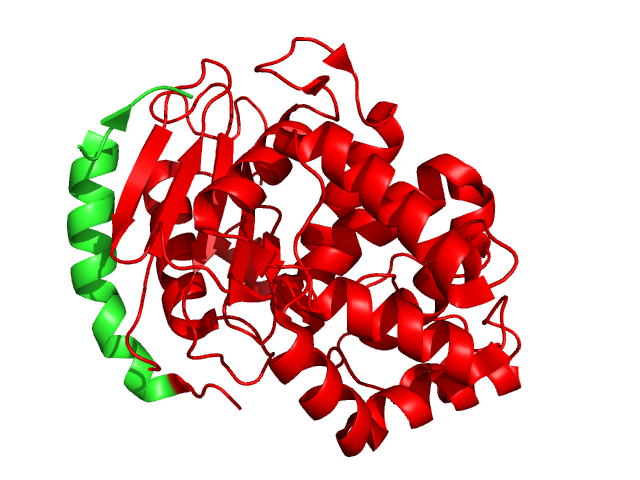
Для удобства здесь представлена только одна цепь, красным выделены остатки, которые входят в домен по Pfam. Видно, что разница не особо принципиальная, но почему в домене Scop лежит вся цепь, а в домене Pfam выкинута одна альфа-спираль и еще один небольшой кусок, я не совсем понимаю…