Для выполнения работы был выбран белковый домен с AC: PF20618 из практикума 11.
Выравнивание проводили с помощью программ: MAFFT, MUSCLE, T-COFFEE. Ниже предоставлен файл Jalview cо сравниваемыми выравниваниями одних и тех же последовательностей
Далее мы сравнили полученные результаты, используя программу Macho.py , сделанную однокурсниками, получили список одинаково выровненных колонок и представили их в таблице 1.
| Mafft и Muscle | Mafft и T-coffee | |
| Блоки одинаково выровненных колонок | (1,26) = (1,26); (45,53) = (44,52) | (1,33) = (1,33); (45,54) = (45,54) |
- Сравнение MAFFT с T-COFFEE показывает более длинные блоки совпадения (33 колонки подряд), чем сравнение MAFFT с MUSCLE (26 колонок).
- Это может говорить о том, что выравнивания MAFFT и T-COFFEE более похожи друг на друга по первой части выравнивания.
- Для MAFFT и MUSCLE второй блок совпадения — 9 колонок (45–53 у MAFFT и 44–52 у MUSCLE.
- Для MAFFT и T-COFFEE — 10 колонок (45–54).
- Это тоже говорит о близости результатов, но с небольшими сдвигами индексов.
- Для блока (45,53) у MAFFT соответствует (44,52) у MUSCLE — на 1 колонку сдвиг.
- Для MAFFT и T-COFFEE индексы совпадают точно.
- MAFFT и T-COFFEE дают более похожие результаты выравнивания, чем MAFFT и MUSCLE.
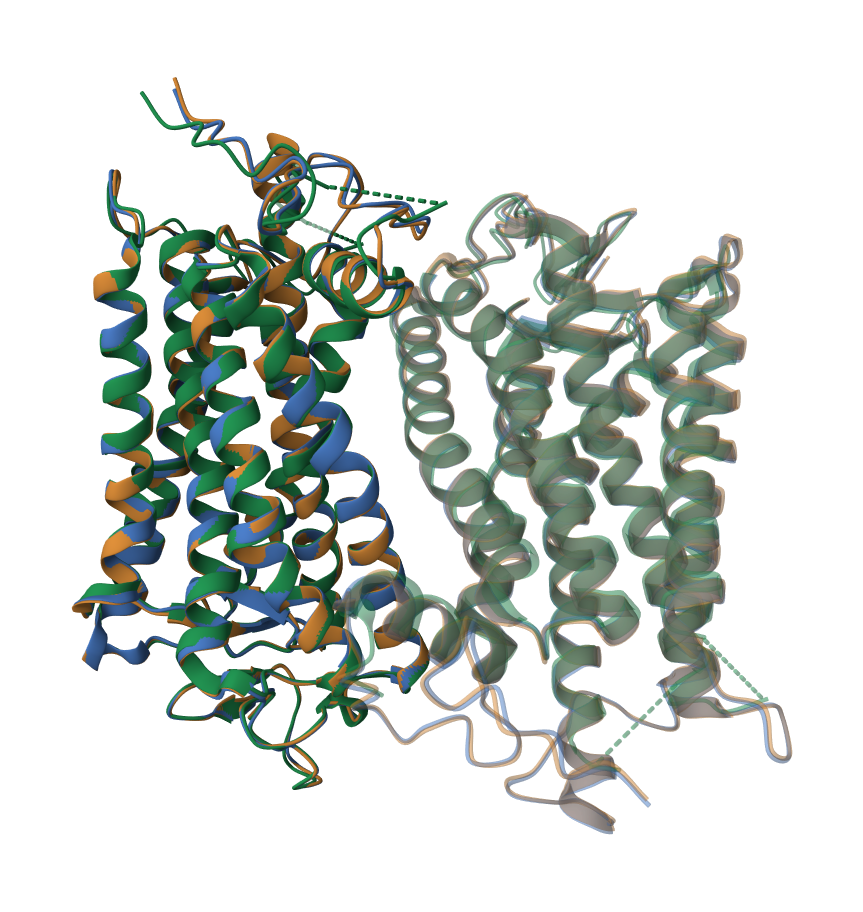
Для выполнения задания выбрали 3 белка семейства rhodopsin из Pfam: 2ped, 2g87, 1f88.
| Muscle и PDB | |
| Блоки одинаково выровненных колонок | (1,236) = (1,236); (238,239) = (238,239); (241,326) = (241,326); (329,331) = (329,331); (335,348) = (335,348) |
| Одинаково выровненные колонки, не входящие в блоки | (333) = (333) |
Muscle (последовательностное выравнивание) и PDB (структурное) дали очень близкие результаты, что говорит о высокой консервативности белков семейства rhodopsin. Расхождения могут быть связаны с: Разными алгоритмами (Muscle учитывает только последовательность, а структурное выравнивание – ещё и 3D-форму). Гибкими участками (например, петлями), которые плохо выравниваются на последовательностном уровне. Участки 241-326 и 335-348 могут включать трансмембранные спирали (родопсины – мембранные белки). Короткие совпадающие блоки (например, 238-239) могут соответствовать ключевым каталитическим или связывающим сайтам.
MUSCLE (Multiple Sequence Alignment) — это популярный алгоритм для множественного выравнивания биологических последовательностей (белков или нуклеотидов), разработанный Робертом Эдгаром в 2004 году.
Ключевые особенности:
Высокая скорость и точность: MUSCLE сочетает итеративные методы и алгоритмы кластеризации для улучшения выравнивания.
Эффективность: Оптимизирован для работы с большими наборами данных (до ~10 000 последовательностей).
Три стадии выравнивания:
Draft alignment: Быстрое грубое выравнивание на основе k-mer.
Improved alignment: Уточнение с помощью итеративных методов.
Refinement: Дополнительная оптимизация для консервативных участков.
Источники:
Edgar, R.C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32(5), 1792–1797. DOI:10.1093/nar/gkh340
.Статья в PubMed: PMID:15034147.