Нам дана оптимизированная структура этана в виде z-matrix :
$DATA eth C1 C C 1 cc H 2 ch 1 cchv H 2 ch 1 cch 3 d1 0 H 2 ch 1 cch 3 d2 0 H 1 ch 2 cch 3 d3 0 H 1 ch 2 cch 5 d3 0 H 1 chv 2 cch 4 d3 0 cc=1.52986 ch=1.08439 chv=1.08439 cch=111.200 cchv=111.200 d1=120 d2=-120 d3=180 $ENDВместо значений длин и углов связей стоят переменные.
Наша цель состоит в том, что бы создать порядка 20 разных файлов для расчёта энергии в Gamess с разными значениями по длине одной из связей. Для этого в представленных координатах одна связь или угол отличается названием переменной, но не её значением.
Попробуем автоматизировать процесс с помощью скрипта на bash. Bash это интерпретатор командной строки, который автоматически запускается при присоединении к kodomo.
Для этого к координатам добавьте шапку для dft из предыдущего практикума:
$CONTRL COORD=CART UNITS=ANGS dfttyp=b3lyp RUNTYP=ENERGY $END $BASIS GBASIS=N31 NGAUSS=6 POLAR=POPN31 NDFUNC=1 $END $GUESS GUESS=HUCKEL $END $system mwords=2 $end $DATAТолько изменим информацию о типе входных координат: COORD=CART на COORD=ZMT.
Проверим, работает ли файл-заготовка. Для этого запустим GAMESS. В результате получаем файл et.log без ошибок. Значит, можно перейти к следующему пункту.
#!/bin/bash
### делаем цикл от -10 до 10 #####
for i in {-10..10}; do
#### нам надо рассчитать новую длину связи #####
#### с шагом 0.02 ангстрема, #####
#### воспользуемся калькулятором bc #####
#### и результат поместим в переменную nb #####
nb=$(echo "scale=5; 1.52986 + $i/50" | bc -l)
#### пролистаем файл et.inp и заменим указание переменной ###
#### на новое значение и пере направим результат в файл ###
sed "s/cc=1.52986/cc=$nb/" et.inp > b_${i}.inp
done
где 1.52986 - стартовая длина изменяемой связи.Запустим скрипт :
bash ./make_b.bashВ результате получаем 21 inp файл, в каждом - разное значение для переменной сс.
doneвставим запуск Gamess:
gms b_${i}.inp 1 > b_${i}.log
Теперь нам надо извлечь значение энергии из log файла. Для этого удобно пользоваться awk. Сначала в нашем скрипте комментируем запуск Gamess, поставив в начало строчки c gms #. Добавим после закомментированной строчки вызов awk, при этом мы ищем строчку с TOTAL ENERGY и печатаем 4ое поле, считая, что поля разделены пробелами:
awk '/TOTAL ENERGY =/{print $4}' b_${i}.log
После запуска скрипта на экране появилось 21 значение энергии. Теперь удобно было бы выводить и значение длины связи, для этого нам нужно добавить перед вызовом awk распечатку переменой nb. Распечатаем переменную и несколько пробелов без переноса строки:
echo -n "$nb "Теперь запускаем скрипт и далее перенаправляем поток в файл : bash ./make_b.bash > bond .
В результате имеем файл bond с длинами связи и соответствующими значениями энергии.
Запускаем Gnuplot:
gnuplotДалее просто введём: plot "bond"
Появился график с точками похожими на параболу. Теперь нам надо найти коэффициенты в функции f(x)=a+k(x-b)^2, которые бы позволили наиболее близко описать наблюдаемую зависимость. Для этого воспользуемся возможностями Gnuplot. Сначала зададим функцию в развернутом виде, в строке gnuplot введём:
f(x)=a + k*x*x - 2*k*x*b + k*b*bИ зададим стартовые значения коэффициентов:
a=-80 k=1 b=1.5Проведём подгонку коэффициентов под имеющиеся точки в файле bond:
fit f(x) "bond" via a,k,bЗначения коэффициентов:
a = -79.7652 +/- 0.0004522 (0.000567%) k = 0.563608 +/- 0.02335 (4.142%) b = 1.55432 +/- 0.002455 (0.1579%)Далее построим графики функции и значений энергии из Gamess:
plot "bond", f(x)
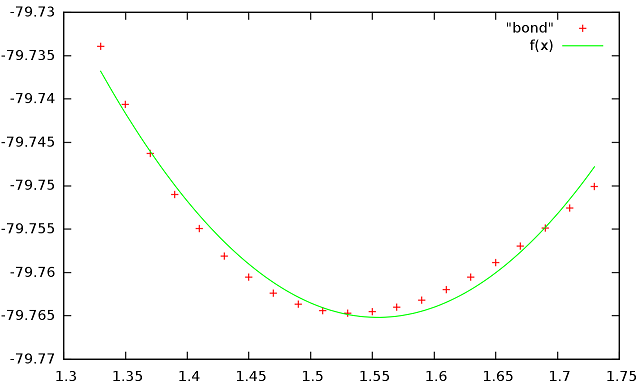
Если посмотреть на коэффициенты, то видно, что самый большой разброс имеет K =>функция и точки не полностью совпадают.
Значения коэффициентов:
a = -79.7647 +/- 1.21e-08 (1.517e-08%) k = 3.56076e-05 +/- 6.229e-09 (0.01749%) b = 111.38 +/- 9.954e-05 (8.937e-05%)
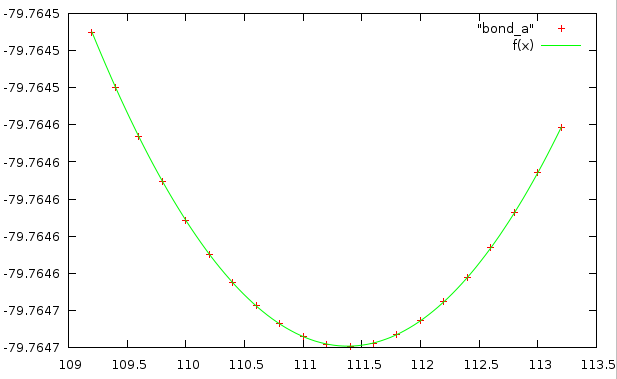
Значения коэффициентов:
Final set of parameters Asymptotic Standard Error ======================= ========================== a = 0.00234519 +/- 8.891e-07 (0.03791%) k = 3.00014 +/- 0.0002247 (0.007491%) b = -79.7623 +/- 6.577e-07 (8.245e-07%)
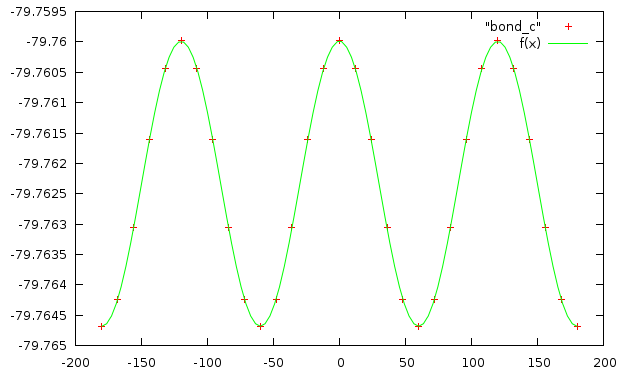
У нас 3 минимума, так как крайние точки совпадают.