На данной странице представлен практикум по анализу вторичной структуры тРНК и ДНК-белковых взаимодействий с помощью программ из пакета EMBOSS и Viena Rna Package.
1) Анализ вторичной структуры тРНК(Arg-tRNA)
В данном упражнении рассматривается комплекс аргинил-тРНК-синтетазы и аргининовой тРНК (PDB ID: 1F7U). Для анализа вторичной структуры тРНК, ее последовательность была выгружена в fasta формате, после чего была запущена программа einverted со стандартными параметрами, кроме Minimum score threshold - 5. Также был реализован алгоритм Зукера программой RNAfold. Полученные результаты сравнивались со значениями, полученными с помощью программы find_pair из практикума 2. Результаты в таблице:
| Участок структуры | Позиции в структуре (find_pair) | Предсказание с помощью einverted | Предсказание с помощью алгоритма Зукера |
|---|---|---|---|
| Акцепторный стебель | 5'-901-907-3' 5'-966-972-3' | - | 5'-901-905-3' 5'-968-972-3' |
| D-стебель | 5'-910-914-3' 5'-922-925-3' | - | 5'-906-912-3' 5'-917-923-3' |
| T-стебель | 5'-949-953-3' 5'-961-965-3' | 5'-949-953-3' 5'-961-965-3' | 5'-942-943-3' 5'-948-949-3' |
| Антикодоновый стебель | 5'-939-944-3' 5'-926-931-3' | - | 5'-936-939-3' 5'-953-956-3' |
| Общее число канонических пар нуклеотидов | 22 | 5 | 18 |
Полученные результаты говорят о низкой предсказательной способности вторичной структуры РНК программой einverted, т.к. предсказаны взаимодействия только из Т-стебля. Возможно это связано с плохо подобранными параметрами программы, но скрипт, написанный мной для их подбора, быстро израсходовал квоту на kodomo... Алгоритм Зукера предсказал больше взаимодействий, но с более низкой точностью, т.к. в контактах не прослеживаются классические взаимодействия, наблюдаемые в тРНК.
2) ДНК-белковые взаимодействия
В данном упражнении рассматривается триптофановый репрессор в комплексе с ДНК (PDB ID: 1TRO).
Для последующей работы с определенными атомами ДНК были определены следующие наборы:
define set1 {*.O?' and dna}
define set2 {*.OP? and dna}
define set3 {nitrogen and dna}
Мной был написн скрипт JMol для визуализации наборов атомов в ДНК. Для запуска скрипта нажмите кнопку "Start", для смены изображения – кнопку "Resume", также Вы можете включить или отключить вращение белка с помощью кнопок "Spin on" и "Spin off" или посмотреть скрипт, нажав на кнопку "Show script".
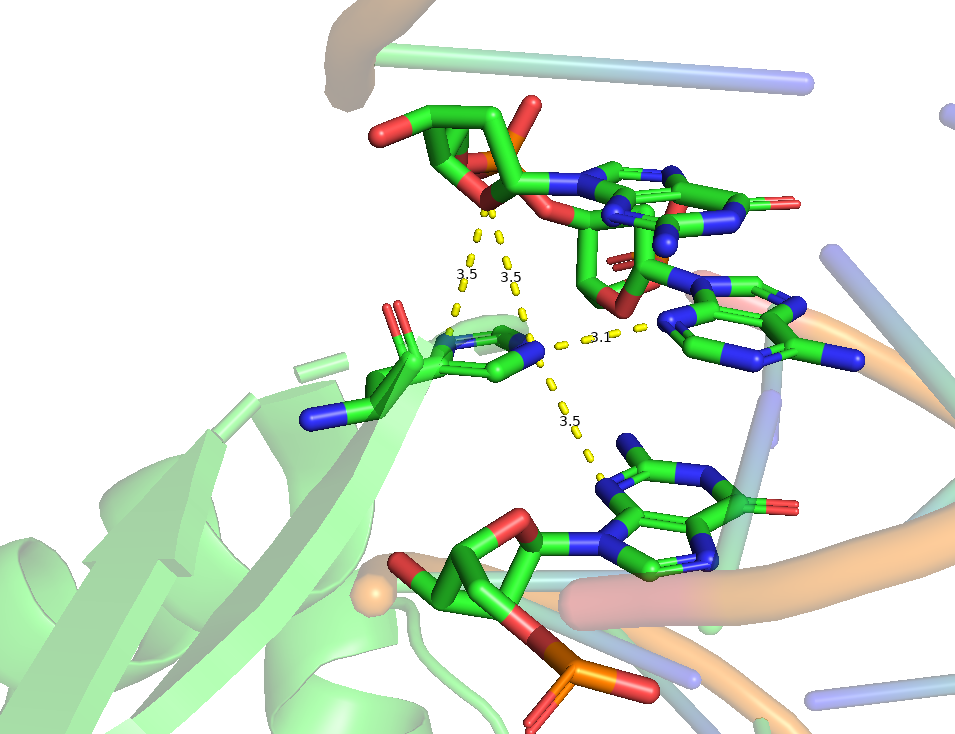
Для исследования ДНК-белковых контактов в комплексе 1TRO, считаем полярными атомы кислорода и азота, контакт между которыми возможен на расстоянии меньшем 3.5 ангстрем, а неполярными - атомы фосфора, углерода и серы, для контакта которых минимальное расстояние - 4.5 ангстрем. Для анализа количества полярных и неполярных взаимодействий мной был написан скрипт, результаты в таблице:
| Контакты белка: | Полярные | Неполярные | Всего |
|---|---|---|---|
| Остатки дезоксирибозы | 4 | 41 | 45 |
| Остатки фосфорной кислоты | 54 | 48 | 102 |
| Остатки азотистых оснований со стороны большой бороздки | 7 | 12 | 19 |
| Остатки азотистых оснований со стороны малой бороздки | 0 | 4 | 4 |
Исходя и указанного в таблице, больше всего контактов с белком(и полярных, и неполярных) наблюдается у остатков фосфорной кислоты. Это вероятно связано с тем, что фосфаты выпетливаются и структуры спирали, что делает упрощает их взаимодействие с другими молекулами. Также интересно отметить тот факт, что у атомов азотистых оснований, обращенных в сторону большой бороздки контактов больше, чем у атомов, обращенных в сторону малой, что связано со стерически более доступными условиями у большой бороздки.
Также с помощью программы nucplot было построено изображение для визуализации ДНК-белковых контактов в молекуле. С рисунком можно ознакомиться по ссылке или ниже:
Аминокислотный остаток, который образует наибольшее количество контактов с ДНК - это His65, вероятно, он и самый важный для связывани, во-первых, из-за количества образуемых связей(4 шт.), во-вторых, из-за способности гистидина участвовать в транспорте протонов.