1. Таксономия и функция нуклеотидной последовательности (из практ. 6)
Сначала я запустила поиск в базе Nucleotide collection программой BLASTX 2.5.1+. Она переводит нуклеотидную последовательность в белковую и ищет сходные белки. Все найденные белки назывались cytochrome c oxidase subunit I. Затем я посмотрела таксономию найденных последовательностей (рис.1а). Видно, что 5 лучших последовательностей относятся к Entoprocta, а три из них - к Loxosomatidae. Однако семейство как уровень таксономии меня не устроило. Поэтому я запустила поиск в базе данных, nr, но только для Loxosomatidae программой BLASTN 2.5.1+, которая ищет сходные нуклеотидные последовательности. На рис.1б таксономия находок. Видно, что из 5 самых лучших находок 3 принадлежат роду Loxosomella, причем Loxosomella varians значительно лучше всех остальных находок.
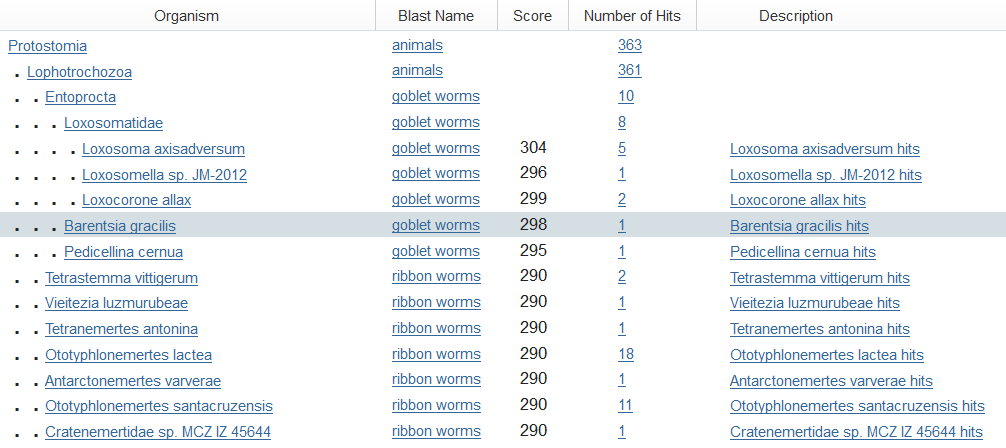 Рис.1а. Часть находок BLASTX, распределенных по таксономии. | 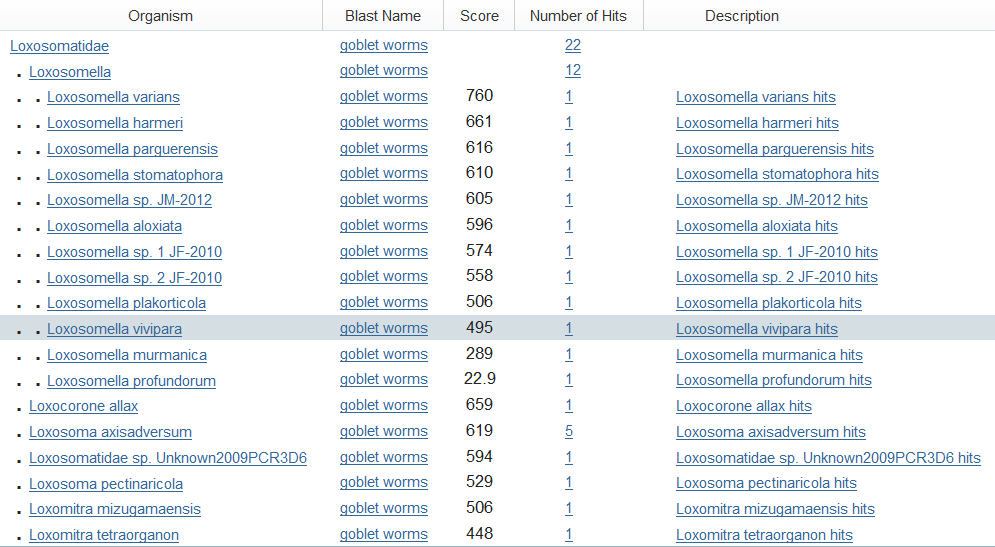 Рис.1б. Часть находок BLASTN, распределенных по таксономии. |
- Protostomia
- blastn - находит похожие, но не обязательно гомологичные последовательности
- discontiguos megablast - находит гомологичные последовательности разных видов.
- megablast - находит гомологичные последовательности близких видов.
3. Наличие гомологов 3 белков в геноме организма.
Мне была дана сборка генома организма Amoeboaphelidium protococcarum, который является внутриклеточным паразитом зеленых водорослей. Я попыталась выяснить, есть ли у него гомологи 3 белков и способны ли они выполнять все свои функции. Для этого я скачала с NCBI последовательности 3 белков в fasta-формате и запустила поиск их гомологов локальным tblastn в базе, созданной из сборки генома. В результате получила 3 файла с выравниваниями для каждого белка.
- HSP7C_HUMAN - белок теплового шока; шаперон; является репрессором
активации транскрипции; учавствует в сплайсинге пре-мРНК.
Файл с выравниванием hsp7c.fasta
Ссылка на файл NCBI HSP7C_HUMAN
В лучшее выравнивание входят 6-612 а.о. белка, которые включают в себя все основные сайты и регионы связывания белков и АТФ. Таким образом, по моему мнению, гомолог белка HSP7C_HUMAN существует и успешно функционирует у Amoeboaphelidium protococcarum.Кол-во выравниваний 16 лучшее выравнивание scaffold-199 score лучшего выравнивания 917 e-value лучшего выравнивания 0.0 positives лучшего выравнивания 89% query cover лучшего выравнивания 94% - TERT_HUMAN - теломераза; восстанавливает длину хромосом при
репликации путем удлинения теломер; связывается с ДНК, с РНК (для обратной транскрипции) и с ионами магния.
Файл с выравниванием tert.fasta
Ссылка на файл NCBI TERT_HUMAN
Лучшее выравнивание включает в себя 452-1007 а.о. При этом сюда не входят два региона связывания РНК (1 – 230 и 325 – 550), регион, отвечающий за специфичность к теломеразной ДНК (137 – 141) и другие не менее важные регионы, а та часть последовательности, которая все-таки входит в выравнивание, обнаруживает очень много несовпадений и даже гэпов. При этом, второе по качеству выравнивание также включает в себя 452-1007 а.о. и очень похоже на первое. По моему, это какой-то другой белок, имеющий некоторое сходство с теломеразой по первичной структуре (вряд ли это гомолог теломеразы, т.к. две копии одного и того же гена, утратившего свои функции, не могли накапливать одни и те же мутации; к тому же, выравнивание слишком плохое, чтобы можно было говорить о гомологии). Надо заметить, что у человеческого белка есть домен обратной транскриптазы 605 – 935 а.о., который входит в выравнивание, но при таком количестве гэпов и несовпадений нельзя сказать, чтобы он выполнял эту функцию. С учетом того, что Amoeboaphelidium protococcarum - это внутриклеточный паразит, который не размножается вне клетки, можно предположить, что он пользуется теломеразой хозяина, когда реплицирует свою ДНК. Тогда ему не нужна ни своя теломераза, ни даже обратная транскриптаза. Вывод - гомологов нет.Кол-во выравниваний 3 лучшее выравнивание scaffold-17 score лучшего выравнивания 105 e-value лучшего выравнивания 8e-23 positives лучшего выравнивания 44% query cover лучшего выравнивания 50% - PABP2_HUMAN - полиаденилат связывающий белок; участвует в форматировании
3' конца пре-мРНК, присоединяя к нему поли-А хвост. Стимулирует создание поли-А хвоста поли-А полимеразой, регулирует его
длину, а затем присоединяет к пре-мРНК. Имеет 2 региона связывания с поли-А полимеразой, домен узнавания РНК.
Файл с выравниванием pabp2.fasta
Ссылка на файл NCBI PABP2_HUMAN
Лучшее выравнивание включает в себя 168-253 а.о. Почти все остальные выравнивания ограничены этими же рамками. Известно, что 172–249 а.о. образуют домен узнавания РНК (RRM). При этом качество выравнивания довольно высоко. Следовательно, можно сказать, что Amoeboaphelidium protococcarum имеет гомолог домена RRM в своем геноме, вероятно, в составе генов других белков. Что же касается функции связывания поли-А хвоста, она, скорее всего, этим организмом утеряна. Трудно сказать, как он справляется без нее, ведь без поли-А хвоста м-РНК в клетке быстро деградирует. Вывод - гомологов нет, зато есть гомологи домена, содержащегося в этом белке.Кол-во выравниваний 16 лучшее выравнивание scaffold-100 score лучшего выравнивания 117 e-value лучшего выравнивания 2e-28 positives лучшего выравнивания 74% query cover лучшего выравнивания 28% 4.Ген белка, закодированный в одном скэффолде Amoeboaphelidium protococcarum.
Для того чтобы получить список скэффолдов с указаниями длин, я использовала команду:
infoseq X5.fasta -only -name -length
В списке я нашла скэффолд длины 38453 - scaffold-700. Затем я получила его последовательность командой:
seqret X5.fasta:scaffold-700 -out scaf700.fasta
Последовательность
Затем я запустила поиск в базе Nucleotide collection программой BLASTN 2.5.1+, ограничив поиск таксоном Opisthokonta (к которому принадлежит в том числе и Amoeboaphelidium). На рис.4а положение находок в скэффолде и статистика по лучшим находкам.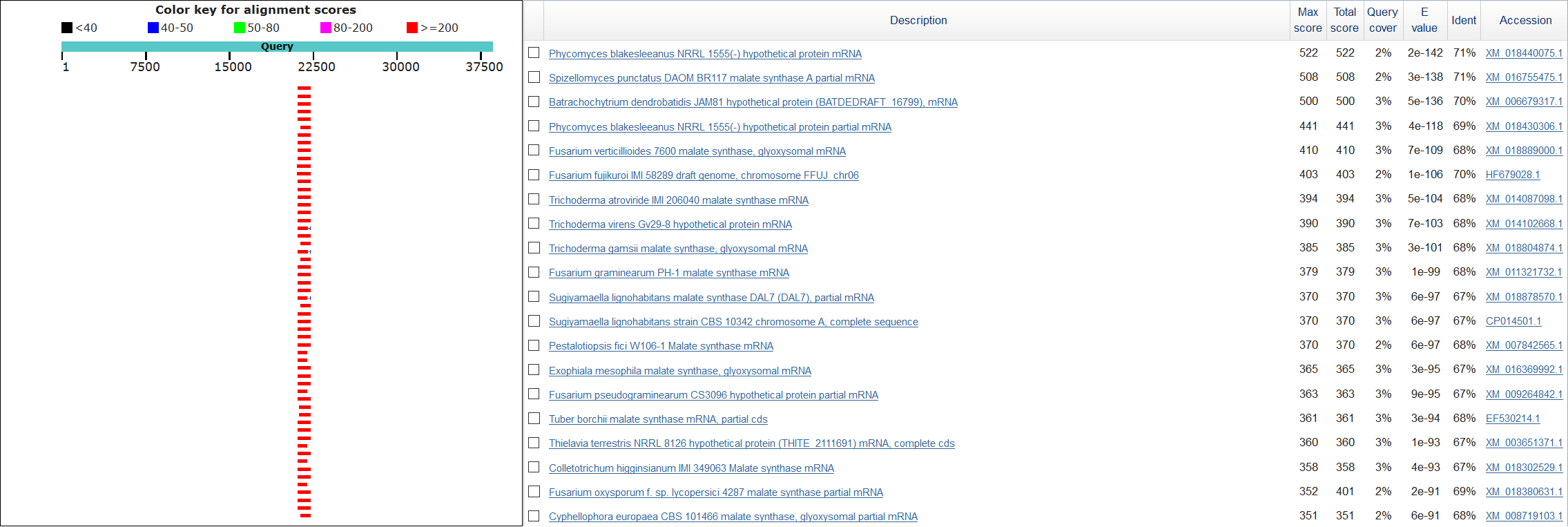
Рис.4а. Фрагменты выдачи BLASTN. Слева положение всех находок в выбранном скэффолде, справа - 20 лучших находок.Видно, что много находок обозначены как мРНК гипотетических белков, но немалое количество являются мРНК малат синтазы. Можно также заметить, что все находки выровнены с одним и тем же местом в скэффолде, что говорит о том, что ген здесь только один. Затем я запустила поиск в базе nr программой BLASTX 2.5.1+ с тем же ограничением. Результаты - на рис.4б
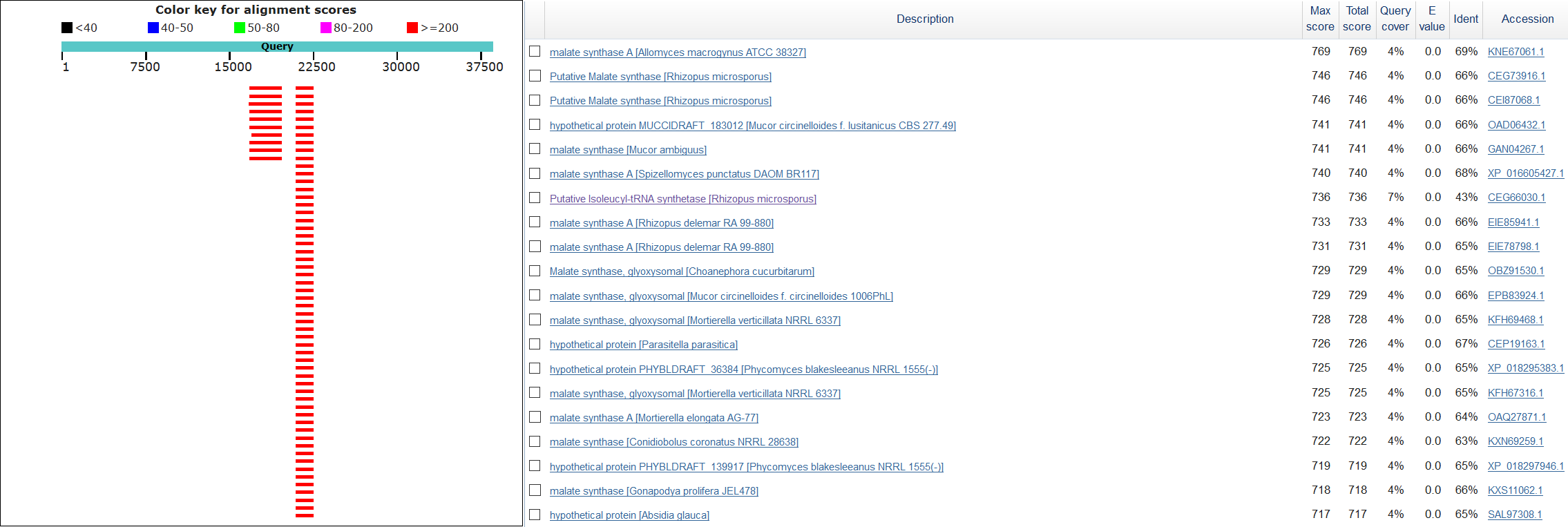
Рис.4б. Фрагменты выдачи BLASTX. Слева положение всех находок в выбранном скэффолде, справа - 20 лучших находок.BLASTX подтверждает предположение о том, что в скэффолде есть ген малат синтазы, однако в транслированной последовательности он обнаружил еще один фрагмент, похожий на последовательность изолейцил-тРНК синтетазы. Тут возможно 2 случая: либо этот фрагмент просто похож на изолейцил-тРНК синтетазу, и гена тут нет, либо ген есть, но blastn не нашел его из-за большого количества более достоверных генов малат синтазы. Чтобы исключить последний вариант, я запустила discontiguous megablast, (чтоб быстрее работал), ограничив поиск образцами, содержащими слова isoleucyl-tRNA synthetase, и задала максимальное число находок 1000. Результат - на рис.4в. Во-первых, положение большинства хороших находок не совпадает с положением, выявленным BLASTX (хотя большое количество маленьких фрагментов расположено в нужном месте); во вторых, все находки довольно-таки некачественные; в третьих, большинство находок - это хромосомы или контиги, а не мРНК; в четвертых, мРНК слишком короткие для того чтобы закодировать всю изолейцил-тРНК синтетазу целиком. Можно предположить, что в этом скэффолде есть какой-то остаток гена изолейцил-тРНК синтетазы, который мутировал до неузнаваемости, либо какой-то эксклюзивный ген, существующий только у этого организма, но, скорее всего, это просто совпадение и гена там нет. Что касается малат синтазы, то ее ген там точно есть. На рис.4г - выравнивание из BLASTX. Гэп в первой строчке - это, скорее всего, интрон. Без него качество выравнивания было бы довольно высоким, но оно и так неплохое. Вывод - в скэффолд-700 входит ген малат синтазы.

Рис.4в. Фрагмент выдачи BLASTN из второго запуска.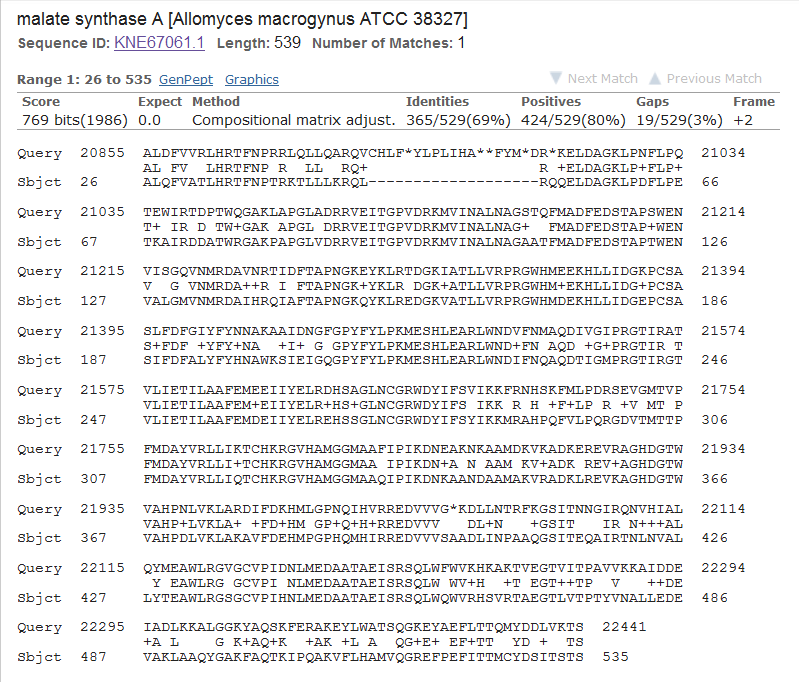
Рис.4г. Выравнивание BLASTX.
- Lophotrochozoa
- Entoprocta
- Loxosomatidae
- Loxosomella varians
2. Сравнение 3-х разных алгоритмов blast
Я запустила поиск в базе Nucleotide collection для той же последовательности, что и в предыдущем задании 3 алгоритмами:
blastn, discontiguous megablast и megablast. Параметры запуска:
Результаты:
| 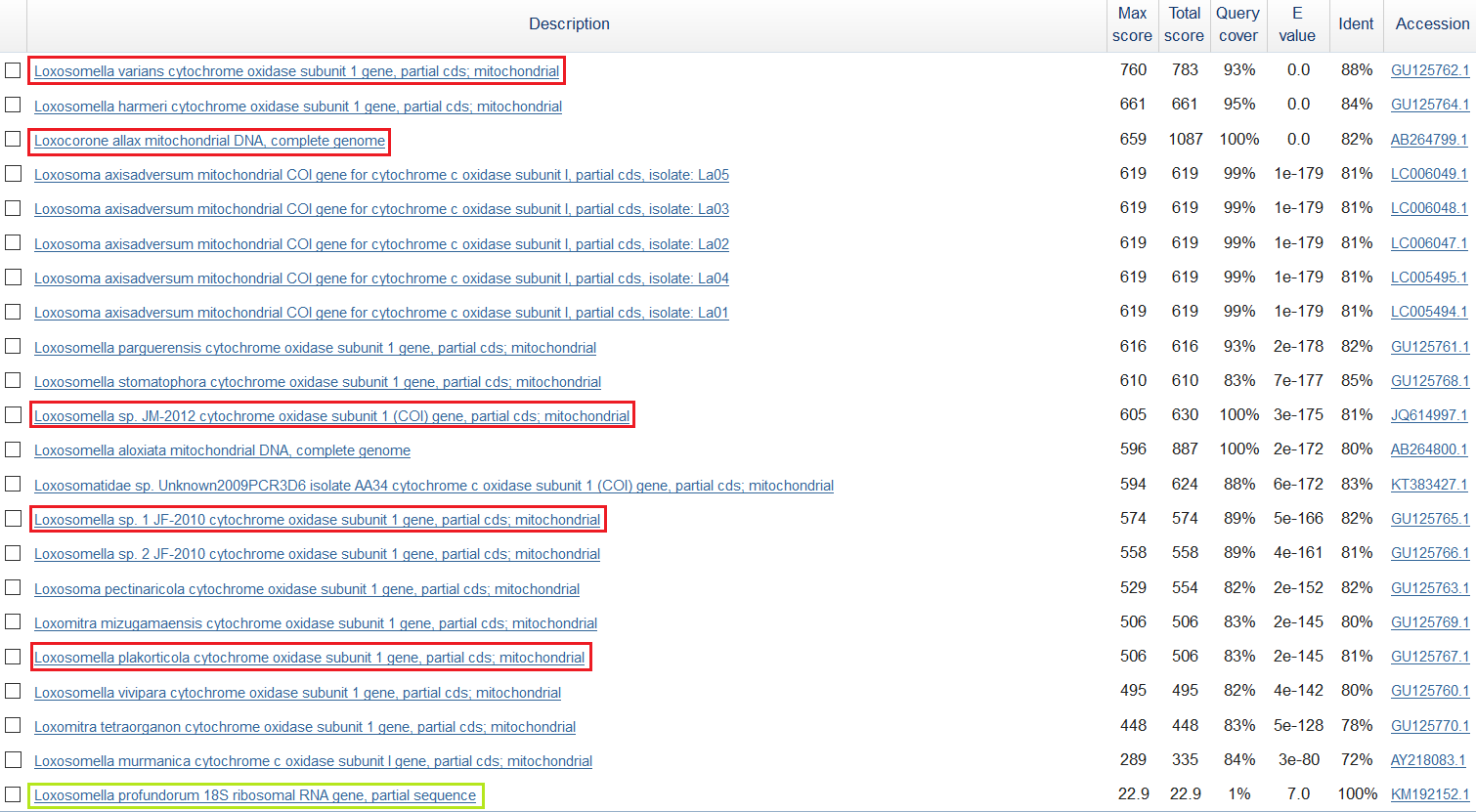 Рис.2a. Находки blastn. Красным обведены все находки megablast, а зеленым - находка, которую нашел только blastn. |
Видно, что почти все находки blastn и discontiguous megablast совпадают, за исключением одной, обладающей весьма плохим e-value и низким query cover. Ее нашел только blastn (рис.2б). Кстати, можно заметить, что она кодирует рибосомную РНК, тогда как прочие, лучшие последовательности кодируют 1 субъединицу цитохром c оксидазы.
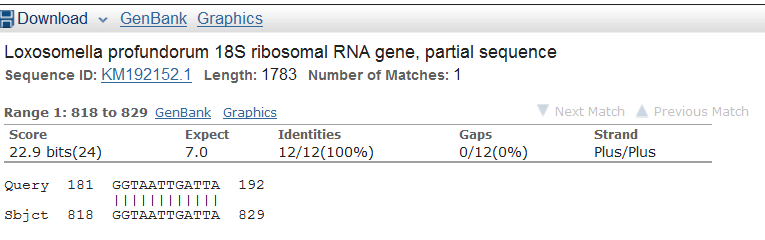
Рис.2б. Находка blastn, которой нет у других алгоритмов.
Помимо этого, можно обнаружить, что, несмотря на одинаковые настройки алгоритмов, total score в одинаковых последовательностях различается. На рис.2в показана выдача по 3 одинаковым последовательностям, и видно, что total score blastn отличается от остальных. Это связано с тем, что blastn сделал несколько выравниваний одной и той же последовательности, которые не были сделаны другими алгоритмами из-за плохих e-value и query cover.
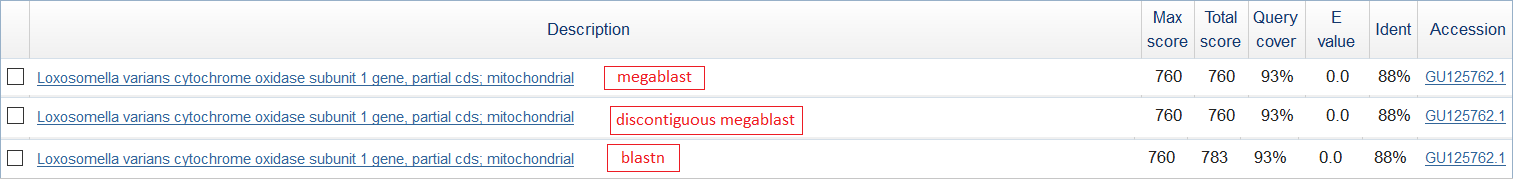
Рис.2в. Статистика по одной находке в разных алгоритмах. Красным цветом
подписаны алгоритмы.
Что касается megablast, то можно увидеть, что 4 из 5 найденных им последовательностей относятся к одному роду Loxosomella, что говорит об их близких родственных связях. На рис.2г изображена последовательность, найденная discontiguos megablast, но не найденная megablast, причем ее score второй по величине в discontiguos megablast. Можно заметить, что в ней нет ни одного участка, совпадающего с моей последовательностью и длиной больше 28.

Рис.2г. Находка discontiguos megablast, которой нет в megablast.
Выводы: