| № | Задание | Команда | Вход | Выход |
| 1 | Несколько файлов в формате fasta собрать в единый файл | echo NC010103.fasta >> list.txt echo NC010167.fasta >> list.txt echo NC010740.fasta >> list.txt echo NC012442.fasta >> list.txt seqret @list.txt out.fasta |
NC010103.fasta NC010167.fasta NC010740.fasta NC012442.fasta list.txt |
out.fasta |
| 2 | Один файл в формате fasta с несколькими последовательностями разделить на отдельные fasta файлы | seqretsplit seqs4.fasta fasta::*.fasta | seqs4.fasta |
nc_010103.1.fasta nc_010740.1.fasta nc_010167.1.fasta nc_012442.1.fasta |
| 3 | Из файла с хромосомой в формате .gb вырезать три кодирующих последовательности по указанным координатам "от", "до", "ориентация" и сохранить в одном fasta файле | echo seqs3.gb:1[2:15] >> list.txt echo seqs3.gb:2[4:20:r] >> list.txt echo seqs3.gb:3[100:125] >> list.txt seqret @list.txt fasta::out.fasta |
seqs3.gb list.txt |
out.fasta |
| 4 | Транслировать кодирующие последовательности, лежащие в одном fasta файле, в аминокислотные, используя указанную таблицу генетического кода. Результат - в одном fasta файле. | transeq human_mito_cds.fasta fasta::out.fasta -table 2 |
human_mito_cds.fasta | out.fasta |
| 5 | Транслировать данную нуклеотидную последовательность в шести рамках. | transeq seq.fasta out.fasta -frame 6 |
seq.fasta | out.fasta |
| 6 | Перевести выравнивание и из fasta формате в формат .msf | seqret fasta::D_AgeI.fasta msf::D_AgeI.msf |
D_AgeI.fasta | D_AgeI.msf |
| 7 | Выдать в выходной поток число совпадающих букв между второй последовательностью выравнивания и всеми остальными (на выходе только имя последовательности и число) | infoalign aln.msf stdout -only -name -simcount | head -n 2 | tail -n 1 | aln.msf |
SplZORFHP_3 27 |
| 8 | Перевести аннотации особенностей в записи формата .gb в табличный формат .gff | featcopy NC_009894.gbk gff::out.gff |
NC_009894.gbk | out.gff |
| 9* | Из данного файла с хромосомой в формате .gb получить fasta файл с кодирующими последовательностями; (*) добавить в описание каждой последовательности функцию белка (из поля product) | extractfeat NC_009894.gbk fasta::out.fasta -featinname 1 -describe product |
NC_009894.gbk | out.fasta |
| 10* | Перемешать буквы в данной нуклеотидной последовательности; (*) проверить с помощью blastn сколько "достоверных" находок (с E-value < 0.1) найдется в нуклеотидном банке данных (запустите с порогом E = 10 - по умолчанию) | shuffle -o out.gbk NC_009894.gbk |
NC_009894.gbk | out.gbk С помощью blastn нашлось 5 "достоверных" (Evalue < 0.1) находок в банке nr/nt, но мин. Evalue=0.019 . |
| 11 | Найти частоты кодонов в данных кодирующих последовательностях | cusp human_mito_cds.fasta codons.cusp | human_mito_cds.fasta | codons.cusp |
| 12 | Найти частоты динуклеотидов в данной нуклеотидной последовательности и сравнить их с ожидаемыми | compseq human_mito_cds.fasta out.compseq -word 2 -calcfreq 1 | human_mito_cds.fasta | out.compseq |
| 13 | Выровнять кодирующие последовательности соответственно выравниванию белков - их продуктов | tranalign cdss_D-ddeI.fasta proteins_D-DdeI.fasta fasta::out.fasta | cdss_D-ddeI.fasta proteins_D-DdeI.fasta |
out.fasta |
2.Сравнение геномов
2а. Я взяла два полных генома бактерий: Bacillus pumilus strain NJ-M2 и Bacillus subtilis subsp. subtilis str. 168 . Для них я построила выравнивание с помощью blast2seq. Параметры: программа BLASTN 2.5.1+; Word size 16; Expect value 10; Match/Mismatch scores 2,-3; Gapcosts 5,2. На рис.1 карта выравнивания. Параметры выравнивания:
| Max score | Total score | Query cover | E-value | Ident |
| 24525 | 1.710e+06 | 53% | 0.0 | 79% |
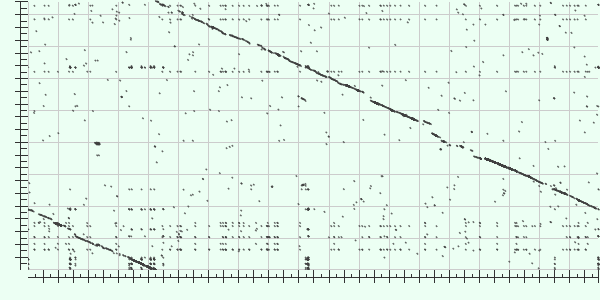
Рис.1. Карта выравнивания.
На рис.2 я обозначила основные эволюционные события. Их было немного. При первом взгляде на карту выравнивания может показаться, что имеет место транслокация. Однако, так как хромосомы кольцевые, это нельзя считать верным. Видно, что выровнялись прямая цепь BACPU и обратная BACSU, однако это не имеет значения, так как понятия прямой и обратной цепи условны. Остальные события - это вставки в геноме BACPU (либо делеции в BACSU) под цифрами 1, 2, 3, 5, 6 в красной рамке, и вставка в геноме BACSU (либо делеция в BACPU) под цифрой 4 в красной рамке. В синей рамке расположено нечто, что я интерпретировала как транслокацию с последовавшей делецией в BACSU.
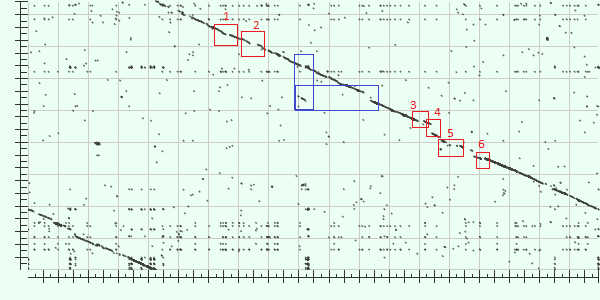
Рис.2. Карта выравнивания с отмеченными эволюционными событиями.
| Номер вставки/делеции | 1(bacpu) | 2(bacpu) | 3(bacpu) | 4(bacsu) | 5(bacpu) | 6(bacpu) |
| Координаты вставки/делеции (приблизительно) | 1300000:1350000 | 1450000:1550000 | 2600000:2650000 | 2100000:2300000 | 2800000:2870000 | 3000000:3050000 |
Я вырезала эти фрагменты из соотв. геномов и поискала их гомологи c помощью megablast в Nucleotide Collection. Это было сделано для того чтобы выяснить, вставка это или делеция, и если вставка, то откуда. События 1, 2, 3, 5, 6 я бы назвала делециями в геноме BACSU. А номер 4, по моему, это вставка в BACSU. На рис.3 распределение находок для №4.

Рис.3. Распределение находок к фрагменту генома BACSU.
Почти все находки относятся к разным штаммам BACSU, однако можно заметить, что большое кол-во находок выровнилось только на первых 50000 нуклеотидах фрагмента. Тем временем, есть одна находка Bacteriophage SPBc2, которая занимает почти всю остальную часть вырезанного фрагмента (Ident=99%). Таким образом, можно предположить, что здесь имела место вставка в геном BACSU из генома Bacteriophage SPBc2 (который специализируется на бактериях рода Bacillus).