Определение сайтов связывания транскрипционного фактора в данном участке хромосомы человека
Мне был дан файл chipseq_chunk3.fastq с ридами Illumina, полученный в результате сhip-seq эксперимента.
1. Анализ качества чтений.
Чтобы проанализировать качество чтений, я применила программу FastQC. Я запустила следующую команду:
fastqc chipseq_chunk3.fastqЯ получила 2 файла: chipseq_chunk3_fastqc.html и chipseq_chunk3.zip. Первый файл - это анализ качества последовательностей в виде html страницы, а второй - архив со служебными файлами для html страницы. Как показалось мне, фильтрация данных не потребовалась, так как чтения оказались достаточно высокого качества (рис.1).
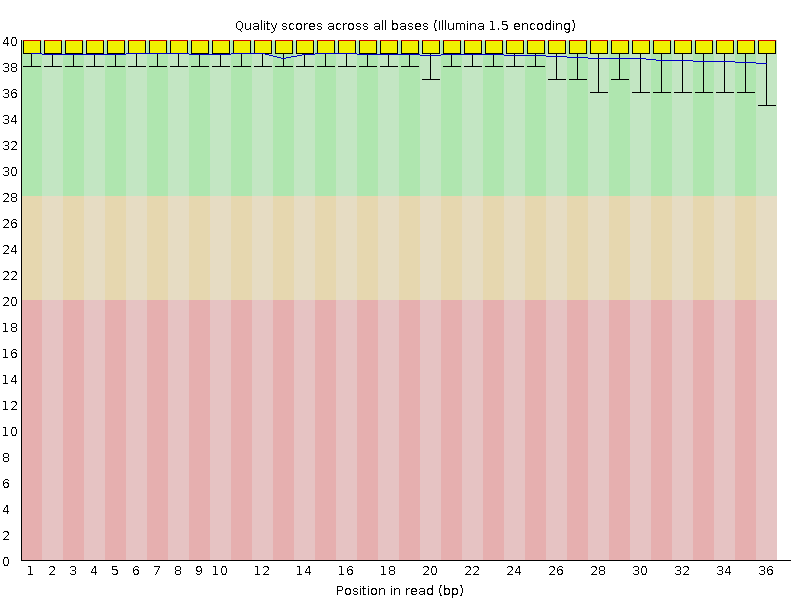
Рис.1. Качество ридов по основаниям.
2. Картирование чтений
Для картирования чтений использовалась программа BWA. Я использовала готовую референсную последовательность генома: GRCh37.p13.genome.fa и соответствующие файлы форматов amb, ann, bwt, pac, sa. Я выровнила мои риды с референсом алгоритмом mem командой:
bwa mem GRCh37.p13.genome.fa chipseq_chunk3.fastq > chipseq_chunk3.samПолучился файл chipseq_chunk3.sam с картированными ридами.
4. Анализ выравнивания
Для перевода файла sam в файл bam использовалась команда:
samtools view chipseq_chunk3.sam -b -o chipseq_chunk3.bamДалее полученный файл chipseq_chunk3.bam был отсортирован по координатам:
samtools sort chipseq_chunk3.bam -T prefix -o sort.bamПолучился файл sort.bam, который я проиндексировала командой:
samtools index sort.bamЭта команда создала файл sort.bam bai. Чтобы выяснить, сколько чтений откартировалось на геном, я сделала так:
samtools idxstats sort.bamЧасть выдачи с хромосомами:
chr1 249250621 33 0 chr2 243199373 27 0 chr3 198022430 27 0 chr4 191154276 22 0 chr5 180915260 22 0 chr6 171115067 20 0 chr7 159138663 18 0 chr8 146364022 16 0 chr9 141213431 15 0 chr10 135534747 22 0 chr11 135006516 23 0 chr12 133851895 6172 0 chr13 115169878 10 0 chr14 107349540 6 0 chr15 102531392 14 0 chr16 90354753 17 0 chr17 81195210 6 0 chr18 78077248 5 0 chr19 59128983 8 0 chr20 63025520 9 0 chr21 48129895 8 0 chr22 51304566 10 0 chrX 155270560 21 0 chrY 59373566 1 0В каждой строчке записаны по порядку: название референса, длина последовательности, кол-во откартированных ридов, кол-во неоткартированных ридов. Как мы видим, откартировались все риды, причем большая часть - на хромосому 12. Можно с уверенностью сказать, что чтения происходят именно с нее.
Поиск пиков
Для поиска пиков использовалась программа MACS. Сначала я запустила ее так:
macs2 callpeak -t sort.bamПрограмма выдала следующее:
WARNING @ Thu, 25 May 2017 20:21:33: Too few paired peaks (0) so I can not build the model! Broader your MFOLD range parameter may erase this error. If it still can't build the model, we suggest to use --nomodel and --extsize 147 or other fixed number instead.То есть пиков нашлось слишком мало (0). Поэтому я запустила ее еще раз вот так:
macs2 callpeak -t sort.bam --nomodelПрограмма выдала 3 файла: NA_peaks.xls, NA_peaks.narrowPeak и NA_summits.bed. Нашлось 6 пиков, все на хромосоме 12. Информация о них - в таблице. Визуализация - на рис.2.
chr start end length abs_summit pileup -log10(pvalue) fold_enrichment -log10(qvalue) name chr12 102099731 102099930 200 102099873 19.00 13.26993 6.71141 6.79030 NA_peak_1 chr12 102187277 102187476 200 102187366 22.00 17.08627 8.09859 10.37925 NA_peak_2 chr12 102610161 102610384 224 102610263 26.00 23.05781 10.30534 16.04899 NA_peak_3 chr12 102696413 102696638 226 102696543 26.00 23.19511 10.38462 16.17530 NA_peak_4 chr12 102834952 102835243 292 102835099 35.00 26.86287 9.57447 19.67171 NA_peak_5 chr12 102868276 102868588 313 102868423 62.00 53.64547 13.63636 44.18949 NA_peak_6
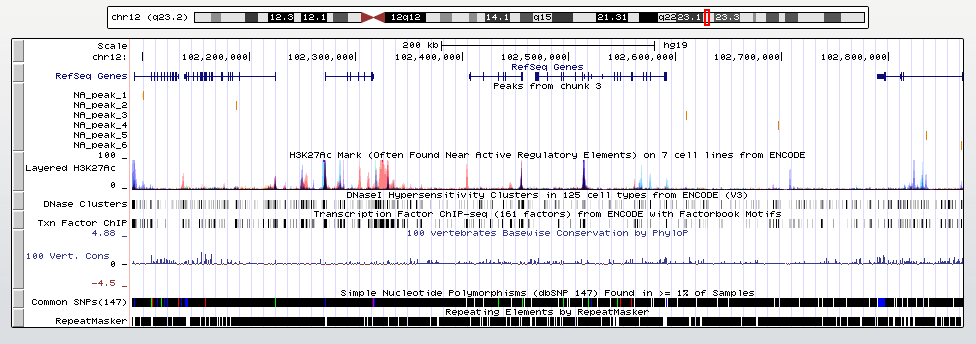
Рис.2. Визуализация пиков в UCSC Genome Browser.
Самый досотверный пик - NA_peak_6 (с лучшими pvalue и qvalue). Его визуализация - на рис.3.
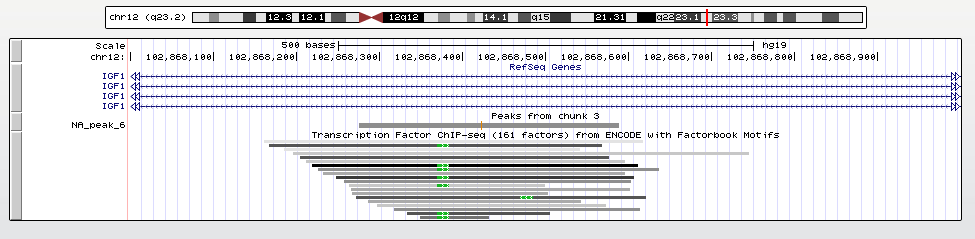
Рис.3. Визуализация NA_peak_6 в UCSC Genome Browser.
Как можно заметить, этот пик расположен прямо на гене IGF1, но едва ли он влияет на его транскрипцию. Можно также заметить, что этот пик подтвержден результатами 20 других chip-seq экспериментов, что лишний раз доказывает его достоверность. Наш пик симметричен, что еще раз подтверждает его правильность.
Выравнивание
Я конвертировала пики в формат fasta с помощью следующей команды:
/srv/databases/ngs/tools/seqtk/seqtk subseq GRCh37.p13.genome.fa NA_peaks.narrowPeak > my_peaks.faПолученные последовательности my_peaks.fa я выровнила программой Muscle. Получилось выравнивание align.fasta.