Ресеквенирование. Поиск полиморфизмов у человека
Анализ качества чтений и их очистка
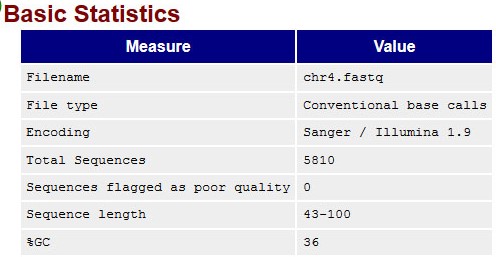
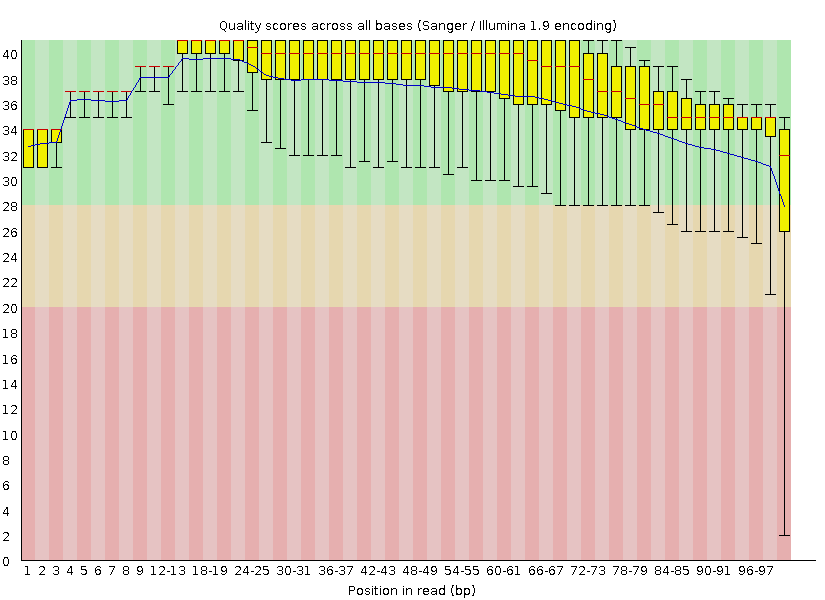
Мне была дана хромосома человека chr4.fasta, а так же файлы с ридами - chr4.fastq
Следующими командами я получил информацию о качестве ридов, а так же очистил их, убрав все нуклеотиды с качеством прочтения меньше 20, и все риды длинной меньше 49 включительно:
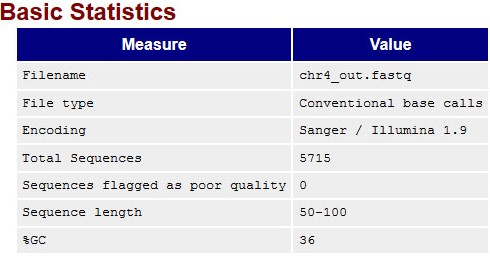
fastqc chr4.fastq java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr4.fastq chr4_out.fastq TRAILING:20 MINLEN:50 fastqc chr4_out.fastq
Результат:
 |
 |
 |
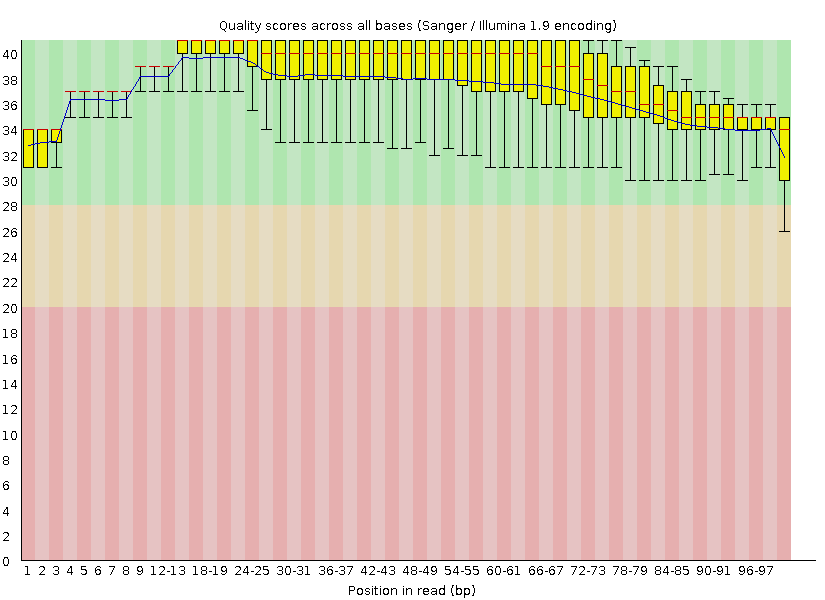 |
По итогу были удалены 95 чтений, а так же минимальная длина поднялась с 43 до 50. Так же были удалены плохо читаемые нуклеотиды на концах чтений - остатки от секвенирования, при котором первые и последние нуклеотиды не расчитываюся на читаемость, в следствии особенности работы ферментов (полимераз).
Картирование чтений
Сначала я проиндексировал референсную последовательность (chr4.fasta)
bwa index chr4.fasta
После этого было посторено выравнивание прочтений и проиндексированного референса с помощью алгоритма mem в формате sam.
bwa mem chr4.fasta chr4_out.fastq > align.sam
Потом с помощью пакета samtools формат sam был переведен в бинарный формат bam. Использовались параматры -b для перевода в bam, и -o для записи в файл.
samtools view align.sam -b -o align.bam
Далее выравнивания чтений с референсом были отсартированы.
samtools sort align.bam -T tmp.txt -o al_sort.bam
Далее файл был проиндексирован. Так же была полученна информация о количестве откартированных ридов.
samtools index al_sort.bam samtools idxstats al_sort.bam > out.txt
Результат - откартировался 5715 рид, то есть каждый прошедчший очистку.
Поиск SNP и инделей
Сначала был создан файл с полиморфизмами в формате bcf с помощью команды:
samtools mpileup -uf chr4.fasta al_sort.bam -o poly.bcf
Потом был создан файл со списком отличий между референсом и чтениями в формате vcf
bcftools call -cv poly.bcf -o diff.vcf
Описание трех полиморфизмов
| Координата | Тип полиморфизма | В референсе | В ридах | Глубина покрытия | Качество ридов |
| 68436883 | SNP | T | C | 37 | 221.999 |
| 88757565 | INDEL | ATGTGTGTGTGTGTGTGTGT | ATGTGTGTGTGTGTGTGTGTGT | 1 | 17.548 |
| 88760642 | INDEL | AAGAGA | AAGAGAGA | 17 | 91.4667 |
Аннотация SNP
Необходимо было проаннотировать полученные SNP, используя базы данных: refgene, dbsnp, 1000 genomes, GWAS, Clinvar.
Для начала необходимо преобразовать файл, чтобы его могла воспринимать основная программа (перед этим я вручную удалил индели, так что файл теперь snp.vtf).
perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp.vcf -outfile snp.pushkin
Далее полученный файл использовался для аннотации SNP с помощью скрипта annotate_variation.pl.
refgene
perl /nfs/srv/databases/annovar/annotate_variation.pl -out chr4.refgene -build hg19 snp.pushkin /nfs/srv/databases/annovar/humandb/
Группы, на которые делит refgene (и количество попавших в них). Всего SNP 42 (при этм Indel'ей было еще плюс 5 ).
| intronic | 36 |
| exonic | 3 |
| downstream | 2 |
| UTR3 | 1 |
| hom | 25 |
| het | 17 |
Мои snp попали в гены: STAP1, MEPE, KLKB1.
dbsnp
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr4.dbsnp -build hg19 -dbtype snp138 snp.pushkin /nfs/srv/databases/annovar/humandb/
1000 genomes
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr4.1kgenomes -build hg19 -dbtype snp138 snp.pushkin /nfs/srv/databases/annovar/humandb/
GWAS
perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -out chr4.gwas -build hg19 -dbtype gwasCatalog snp.pushkin /nfs/srv/databases/annovar/humandb/
Здесь анотированны полиморфизмы, ассоциированные с риском возникновения некоторых болезней. Так, на пример:
Parkinson's disease Cardiovascular disease risk factors Metabolite levels Obesity-related traits
Clinvar
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr4.clincar -buildver hg19 -dbtype clinvar_20150629 snp.pushkin /nfs/srv/databases/annovar/humandb/
Особенность этой базы данных, в том, что в ней находятся записи, связаннывающие полиморфизмы с различными патогенами. Так на пример, у меня нашлась такая одна запись:
clinvar_20150629 CLINSIG=pathogenic;CLNDBN=Prekallikrein_deficiency;CLNREVSTAT=no_assertion_criteria_provided;CLNACC=RCV000012817.24;CLNDSDB=MedGen:OMIM:SNOMED_CT;CLNDSDBID=C0272339:612423:48976006 chr4 187158034 187158034 G A hom 221.999 83
| на главную |
© Гавриш Глеб 2016 |