9.Совмещение структур
Работая с все тем же белком с PDB ID: 5TYB я воспользовался сервисом PDBeFold для нахождения гомологичных структур. Т.к. в моей структуре лишь 1 цепь аминокислотная, а 3 другие представляют собой лишь синтетическую ДНК, поиск я вел только по цепи А. В результате я отобрал 5 структур с RMSD между 0,8 и 2,5 и длиной выравнивания более 50% от длины моего белка. Список выбранных структур приведен в таблице 1.
| PDB ID | Описание |
| 4lzd | HUMAN DNA POLYMERASE MU- APOENZYME |
| 2ihm | POLYMERASE MU IN TERNARY COMPLEX WITH GAPPED 11MER DNA DUPLEX AND BOUND INCOMING NUCLEOTIDE |
| 4qzf | MOUSE TDT, F401A MUTANT, IN COMPLEX WITH A DSB SUBSTRATE AND ZN2+ |
| 4i2h | TERNARY COMPLEX OF MOUSE TDT WITH SSDNA AND AMPCPP |
| 5d46 | STRUCTURAL BASIS FOR A NEW TEMPLATED ACTIVITY BY TERMINAL DEOXYNUCLEOTIDYL TRANSFERASE: IMPLICATIONS FOR V(D)J RECOMBINATION |
После этого, этим же сервисом было построеено множественное выравнивание, которое можно скачать по ссылке. На рис. 1 представеленна визуализация 3D выравнивания.
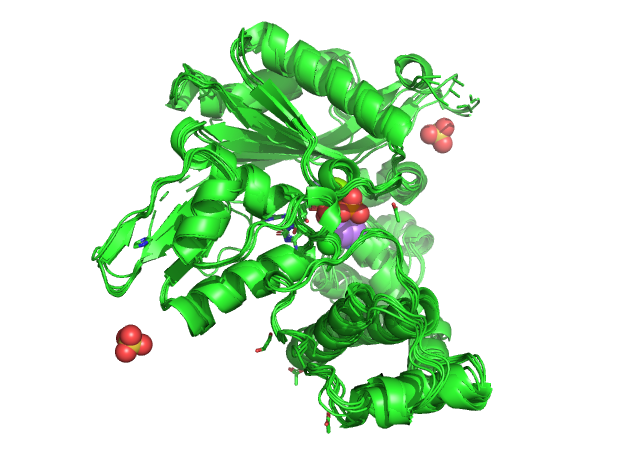
Рисунок 1. Результат работы сервиса PDBeFold.
Также я построил выранвавние сервисом Muscle. Сравнив эти два выравнивания я не обнаружил значительных отличий - все кластеры примерно совпадают. Однако мне удалось найти одно интересное место в позиции 328 (рис. 2), в котором в выравнивании сервиса PDBeFold имеется интересное отличие - аргинины (R) отделенны гепами так, что формируют колонку вместе с глутаминовой кислотой. Это интересно, т.к. часто вводят штрафы за открытие гепа, что в случае верхнего выранивания происходит 2 раза, по сравнению с 1 для Muscle.

Рисунок 2. Фрагмент выравниваний сервисом и (сверху) и Muscle (внизу). Интересующая нас позиция - 328 (нумерация по верхнему выравниванию).
Однако при взгляде на расположение остатка в трехмерной структуре (рис. 3) становиться понятно, что все аимнокислоты в данном столбике находятся в одном месте петли пептида. Также, если заметить что обе аминокислоты (R и E) в норме имеют заряд (+ и - соответсвненно), возникает мысль, что в данном месте может возникать контакт с другой молекулой (например с другим белком) с образованием солевого мостика. Т.е. для отбора не столько важен знак заряда, сколько важен сам факт наличия заряженной аминокислоты. Этот пример демонстрирует, что в данном случае более информативно скорее выравнивание PDBeFold.
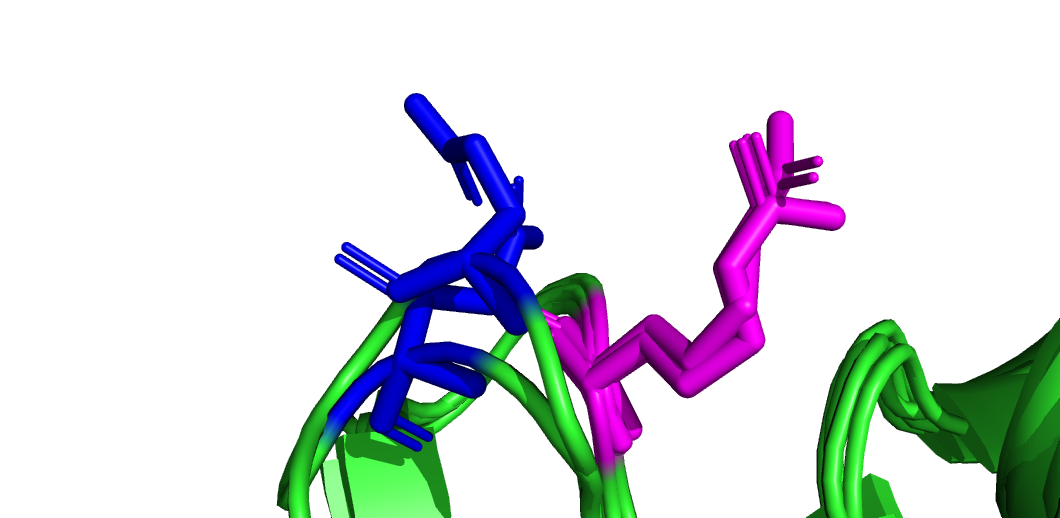
Рисунок 3. Позиция найденная в выравнивании, визуализированная в Pymol. Интересующие остатки E показанны синим, а R - фиолетовым.
| на главную |
© Гавриш Глеб 2017 |