Парное выравнивание. Поиск по сходству
Для выполнения данной работы я использовала последовательность белка фенилацетат-коэнзим А лигазы из генома бактерии Bacteroides Thetaiotaomicron (NP_809341.1) и сервис BLASTP на сайте NCBI.
Задание 1. Собрание выборки гомологов белка phenylacetate-coenzyme A ligase при помощи программы BLASTP из базы Refseq.
Я зарегистрировалась на сайте NCBI, вошла в личный кабинет, после чего запустила программу Protein BLAST
(поиск схожих участков в последовательностях белков) по алгоритму blastp (protein-protein BLAST) для базы данных Refseq (refseq_protein). При использовании параметров
по умолчанию (100 последовательностей на выходе без таксономического ограничения), оказалось, что у данного белка в базе очень много гомологов. В первой сотне у
самой худшей находки E-value составлял 0.0, при выдаче тысячи самый плохой показатель E-value был равен 8e-128. Следовательно, необходимо было ограничить поиск
таксоном и запросить больше последовательностей. Ограничение по таксону Bacteria не принесло особых результатов (так как было найдено более 15000 последовательностей),
ведь исследуемый белок бактериальный, но ограничение в рамках Bacteroidetes позволило сократить выборку до 762 последовательностей, включая исходную. Среди них
есть 522, гомологичные вышеупомянутому белку по всей длине (query cover > 80%).
Ссылка на файл стратегии поиска
Рассмотрим некоторые характеристики 3 находок blastp (см. таблицу 1):
| Параметр/Находка | Лучшая | Средняя | Худшая |
| Название последовательности, организм | phenylacetate-CoA ligase [Bacteroides thetaiotaomicron] | phenylacetate--CoA ligase [Vitellibacter vladivostokensis] | MULTISPECIES: branched chain amino acid aminotransferase [Elizabethkingia] |
| Длина выравнивания | 435 | 429 | 564 |
| Bit Score | 899 bits (2322) | 181 bits (459) | 34.7 bits (78) |
| Процент идентичных остатков | 433/435 (99%) | 127/415 (31%) | 61/282 (22%) |
| Процент сходных остатков | 434/435 (99%) | 210/415 (50%) | 107/282 (37%) |
| E-value | 0.0 | 2e-50 | 8.6 |
| Выравнивание из blastp (в формате fasta) | pr11_best.fasta
[просмотр] | pr11_neutral.fasta [просмотр] |
pr11_worst.fasta [просмотр] |
Если воспользоваться условным критерием определения гомологичной последовательности (E-value < 1e-3 и
Query cover >= 70%), то можем насчитать 521 гомолог.
Выборка из 27 последовательностей
Задание 2. Построение множественного выравнивания последовательностей из полученной выборки.
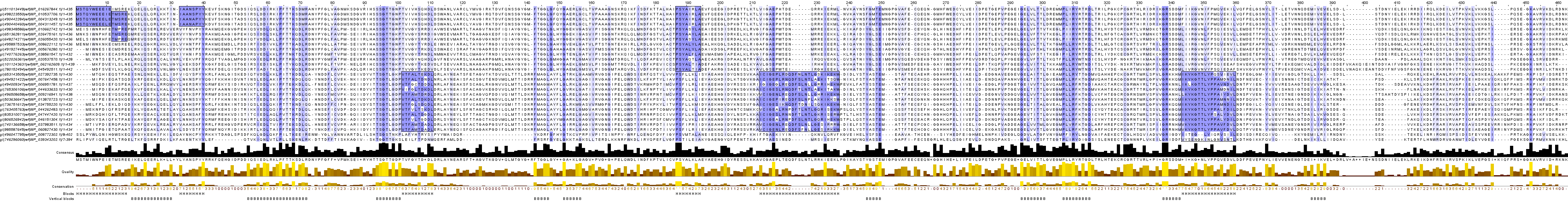
Требовалось скачать последовательности в .fasta и с помощью JailView построить множественное выравнивание
алгоритмом Muscle с параметрами по умолчанию (BLOSUM62 с порогом по консервативности 30%, были удалены невыровненные N- и C-концы). Вертикальные блоки, удовлетворяющие использованному ранее
техническому определению, отмечены символом "B". Приведены примеры блоков, объединяющих
значительное количество последовательностей (но не все), обозначеных символом "H". Количество блоков обоих видов позволяет с большой долей вероятности говорить
о гомологии в отмеченных участках.
Ссылка на проект Jalview
Задание 3. Глобальное и локальное выравнивание исследуемого белка и худшей находки из выборки.
При выполнении задания использовались команды needle и water на сервере kodomo. Выравнивания построены для белков со следующими идентификаторами: WP_008760705.1 и WP_039343202.1. На вход программа требует две последовательности в fasta-формате, величины штрафов за первый гэп (по умолчанию 10.0) и за продление гэпа (по умолчанию 0.5). Needle строит оптимальное глобальное выравнивание двух последовательностей по алгоритму динамического программирования Needleman-Wunsch, основанному на подсчете веса каждого выравнивания (используя матрицу весов и значения штрафов за гэпы, которые определяются пользователем), а затем изучения каждого выравнивания с целью выбрать наилучшее. Выходной файл в fasta-формате можно получить, используя функцию -aformat fasta. Water строит локальные выравнивания (выравнивания неполных последовательностей, имеющие максимальный счет). По умолчанию needle выдает файлы формата .needle, а water соответственно .water, содержащие выравнивание с аннотацией (указывано не только прямое совпадение, но и сходство аминокислот, которые могут быть отмечены одной или двумя точками).
Ссылка на проект с 4 окнами


Задание 4. Выравнивание различных выравниваний друг относительно друга.
Добавлением гэпов было получено выравнивание выравниваний относительно друг друга. В целом достаточно схожи между собой локальные выравнивания, а те участки, которые были выравнены программами по-разному, показывают различие в их алгоритмах. Пример же участка (170-180) с неодинаковым выравниванием можно видеть на рисунке ниже. Интересен тот факт, что на данном примере видно: различаются как глобальные (2 верхних), так и локальные (2 нижних) выравнивания на участке 170-180. Больше всего совпадений наблюдалось в множественном выравнивании и выравнивании, полученном с помощью BLAST.

Ссылка на проект (выравнивание выравниваний)
Задание 5. Парные выравнивания последовательностей двух заведомо негомологичных белков
В пару к своему белку я взяла случайный белок из таблицы: Deoxyribodipyrimidine photo-lyase, Agrobacterium fabrum str. C58 (RefSeq ID: NP_354235.1) и провела те же операции, что в заданиях 3 и 4 (построила выравнивания в needle и water и выровняла их в Jalview друг относительно друга). Выравнивания, полученные разными программами, в большей степени не совпадают. Это связано с большим числом гэпов и маленьким количеством консервативных колонок (наблюдаются только в позициях 400-450). Следовательно, выравнивания недостоверны и по ним нельзя делать выводы о гомологичности анализируемых белков.

Ссылка на проект (выравнивание выравниваний негомологичных белков)
Источники:
[1] База знаний по биологии человека[2] Руководство к JalView
[3] www.bioinformatics.nl/emboss-explorer/
[4] UniProt