Ресеквенирование. Поиск полиморфизмов у человека.
В данном практикуме мной были использованы файлы chr8.fasta, chr8.fastq. Для работы была создана папка
/nfs/srv/databases/ngs/gladmarine.
Я пользовалась FastQC, установленной на kodomo, но для удобства установила также версию с графическим интерфейсом (Java-application).
По результатам работы программы можно сказать, что качество последовательности неплохое, за исключением 3 критериев, которые нужно проверить посредством очистки: Per base sequence content, Per sequence GC content, Sequence length distribution.
Адаптеры в исследуемой последовательности были удалены заранее. Чтобы обрезать с конца чтений нуклеотиды с качеством менее 20, был указан параметр TRAILING:20. С помощью MINLEN:50 были исключены чтения длиной меньше 50 нуклеотидов. В результате работы программы были удалены 140 ридов из всего 8367, таким образом число чтений после чистки составило 8227.
Аналогично полученный файл был проанилизирован программой FastQC.
Используя программу BWA, установленную на kodomo, я откартировала чтения при помощи нижеуказанных команд:
В итоговом файле chr8_snp.vcf описано 100 полиморфизмов. Встречаются как индели, так и замены. Примерно 2/5 от общего числа полиморфизмов имеют хорошее качество прочтения. Глубина покрытия превышает показатель в 50 достаточно редко.
Были произведены аннотации SNP по следующим базам данных - refgene, dbsnp, 1000 genomes, GWAS и Clinvar посредством скрипта annotate_variation.pl. В ANNOVAR существуют 3 типа аннотаций, основанных на:
1) gene-based annotation - генной разметке;
2) region-based annotation - разметке других регионов генома;
3) filter-based annotation - фильтрации.
*Для каждой базы указан тип использованной аннотации.
1. Аннотация по базе refgene (gene-based).
В первой колонке файла *.variant_annotation отображены категории, указывающие местоположение полиморфизма (в скобках указано число снипов данной категории, встретившихся в моем файле):
2. Аннотация по базе dbsnp (filter-based).
76 полиморфизмов имеют rs-идентификатор, соответственно 18 - не имеют. Интересно отметить, что максимальное покрытие в случае snp без rs составило 5, у большинства этот показатель вообще составляет 1 или 2, в отличие от snp из файла *_dropped.
3. Аннотация по базе 1000 genomes (filter-based).
По-прежнему, 76 полиморфизмов имеют указанный идентификатор, частота варьирует от примерно 0,005 до 1.
4. Аннотация по базе Gwas (region-based).
По данным файла можно видеть, что клиническое значение аннотировано в четырёх snp (это болезнь Альцгеймера, регуляция уровня мочевой кислоты, HDL ("хороший") cholesterol), причем среди них нет ни одного экзонного. Следовательно, можно сделать вывод о сложности процессов регуляции и экспрессии генов с данным клиническим значением.
5. Аннотация по базе Clinvar (filter-based).
По результатам работы последней программы, не было найдено аннотаций в базе Clinvar (файл *_dropped оказался пустым).
Часть I. Подготовка чтений.
1. Анализ качества чтений.
FastQC предоставляет возможность проведения простого контроля качества "сырых" последовательностей, полученных в результате высокопроизводительного секвенирования. Программа при помощи ряда статистических анализов позволяет оценить, есть ли в последовательности "проблемы", о которых Вы должны быть осведомлены при дальнейшем анализе. Основые функции следующие:
|
Я пользовалась FastQC, установленной на kodomo, но для удобства установила также версию с графическим интерфейсом (Java-application).
| Использованная команда | Результат |
fastqc chr8.fastq | chr8_fastqc.zip fastqc_report.html |
По результатам работы программы можно сказать, что качество последовательности неплохое, за исключением 3 критериев, которые нужно проверить посредством очистки: Per base sequence content, Per sequence GC content, Sequence length distribution.
2. Очистка чтений.
Trimmomatic позволяет обрезать последовательности адаптеров Illumina (спаренных и неспаренных концевых участков). Выбор режима тримминга и ассоциированных параметров осуществляется в командной строке. Доступные команды:
|
Адаптеры в исследуемой последовательности были удалены заранее. Чтобы обрезать с конца чтений нуклеотиды с качеством менее 20, был указан параметр TRAILING:20. С помощью MINLEN:50 были исключены чтения длиной меньше 50 нуклеотидов. В результате работы программы были удалены 140 ридов из всего 8367, таким образом число чтений после чистки составило 8227.
| Использованная команда | Результат |
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr8.fastq chr8_trimmomatic.fastq TRAILING:20 MINLEN:5 | chr8_trimmomatic.fastq |
Аналогично полученный файл был проанилизирован программой FastQC.
| Использованная команда | Результат |
fastqc chr8_trimmomatic.fastq | chr8_trimmomatic_fastqc.html |
| Сравнение результатов до и после работы с Trimmomatic |
|---|
| Per base sequence quality |
| До очистки чтений |
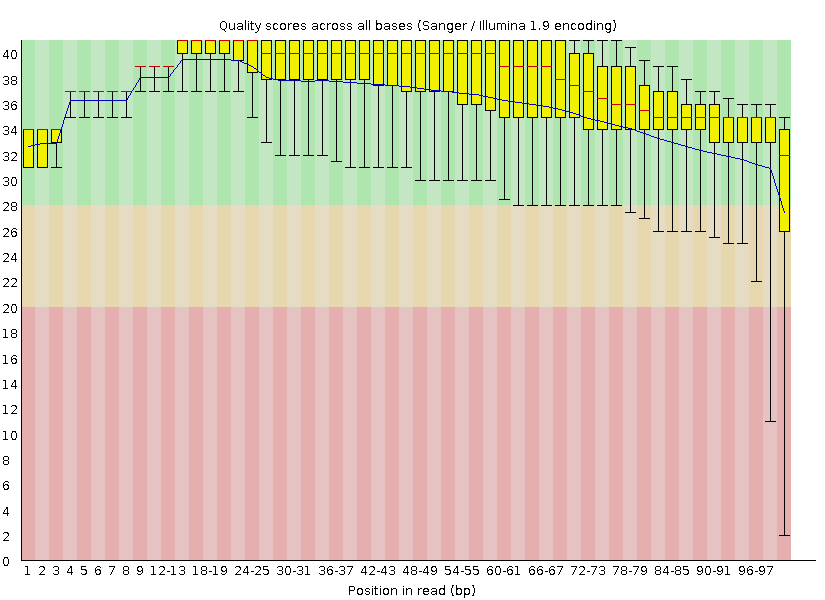 |
| После очистки чтений |
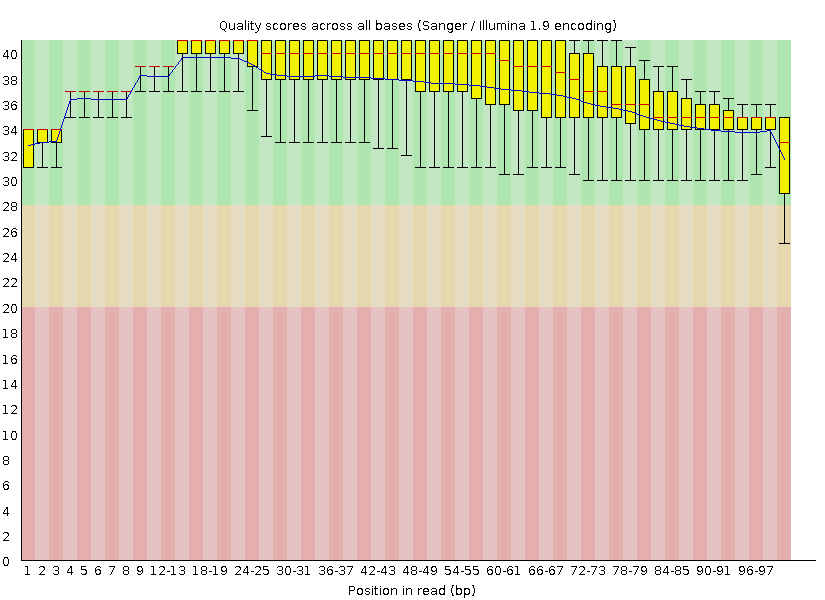 |
| Можно сказать, что Per base quality было достаточно хорошим и до чистки. После работы Trimmomatic не осталось усов, выходящих за пределы оранжевой области. Почти не изменилось значение медианы, и немного увеличилось математическое ожидание. Trimmomatic отбросил чтения низкого качества на конце. |
| Per sequence GC content |
| До очистки чтений |
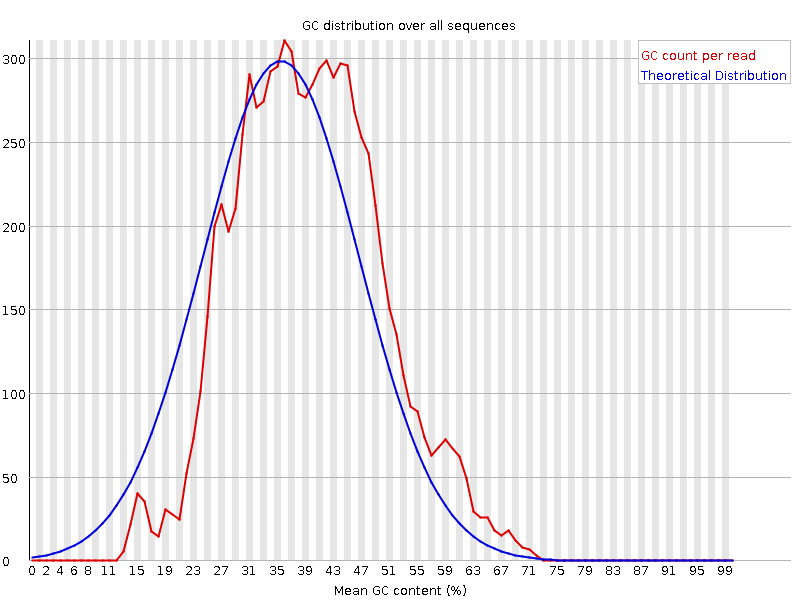 |
| После очистки чтений |
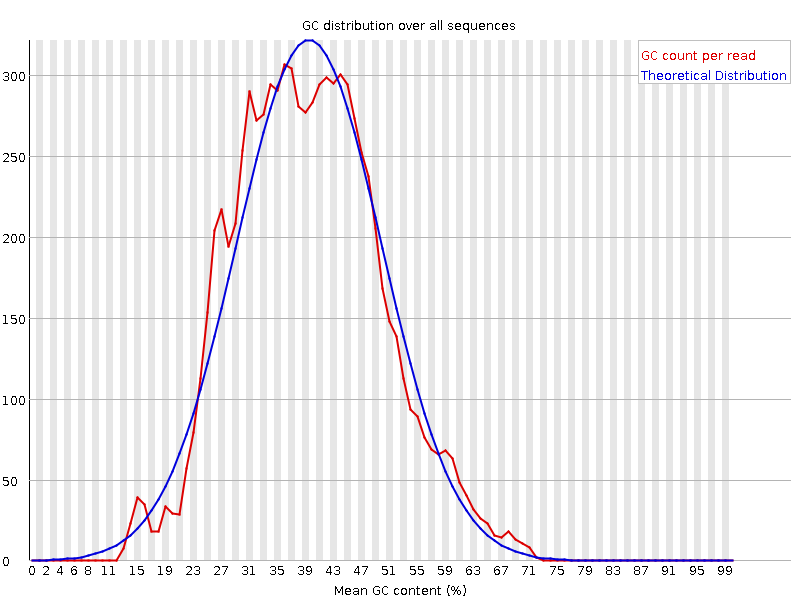 |
| Диаграммы данного модуля были мне интересны тем, что после очистки чтений изменился его "статус". В первом случае возникает предупреждение (!), то есть отклонения составляют более 15%, что хорошо заметно на графике. График не имеет гладкого пика и даже немного сдвинут вправо относительно графика нормального распределения. После работы Trimmomatic Warning не появляется (✓), мы видим, что красный график хорошо накладывается на синий. Правая часть полученного графика становится более гладкой. Наблюдаемые расширенные пики могут свидетельствовать о присутсвии различных загрязнителей. |
Часть II. Картирование чтений.
3. Картирование чтений.
Burrows-Wheeler aligner (BWA) - пакет программного обеспечения для картирования низкодивергирующих последовательностей при сравнении с большими референсными геномами (например, человеческим). Состоит из 3 алгоритмов: BWA-backtrack, BWA-SW и BWA-MEM. Первый разработан для анализа чтений Illumina длиной до 100 п.о., в то время как остальные два оптимизированы для работы с более длинными последовательностями от 70 п.о. до миллиона. BWA-MEM и BWA-SW обладают сходными характеристиками (long-read support и split alignment), но BWA-MEM, самый новый, соответственно наиболее быстрый и точный, рекомендуется для анализа высококачественных последовательностей. BWA-MEM более хорош в использовании при исследовании чтений Illumina 70-100 п.о. (нежели BWA-backtrack). Программа BWA использует преобразование Барроуза — Уилера. [2] |
Используя программу BWA, установленную на kodomo, я откартировала чтения при помощи нижеуказанных команд:
| Использованная команда | Описание | Результат |
bwa index chr8.fasta | Индексирование референсной последовательности | Индексированный chr8.fasta |
bwa mem chr8.fasta | Выравнивание очищенных прочтений и референса с использованием алгоритма BWA-MEM | chr8.sam |
4. Анализ выравнивания.
Для выполнения задания использовался пакет Samtools. [3]| Использованная команда | Описание | Результат |
samtools view chr8.sam | Перевод выравнивания чтений с референсом в бинарный формат .bam. Параметр -b меняет начальный формат на .bam, -o направляет output в файл | chr8.bam |
samtools sort chr8.bam -T | Сортировка выравнивания чтений с референсом (полученный bam-файл) по координате в референсе начала чтения | chr8_sort.bam |
samtools index chr8_sort.bam | Индексирование отсортированного bam-файла | Индексированный chr8_sort.bam |
samtools idxstats chr8_sort.bam | Подсчет числа чтений, откартированных на геном | chr8_result.out |
Часть III. Анализ SNP.
5. Поиск SNP и инделей.
При выполнении использовались также программы пакета BCFtools. [4]| Использованная команда | Описание | Результат |
samtools mpileup -uf chr8.fasta | Создание файла с полиморфизмами в формате .bcf | chr8_snp.bcf |
bcftools call -cv chr8_snp.bcf | Создание файла со списком отличий между референсом и чтениями в формате .vcf | chr8_snp.vcf |
В итоговом файле chr8_snp.vcf описано 100 полиморфизмов. Встречаются как индели, так и замены. Примерно 2/5 от общего числа полиморфизмов имеют хорошее качество прочтения. Глубина покрытия превышает показатель в 50 достаточно редко.
| Примеры полиморфизмов | |||||
|---|---|---|---|---|---|
| Координата | Тип полиморфизма | Референс | Рид | Качество прочтения на участке | Глубина покрытия на участке |
| 116654469 | Инсерция | C | CATA | 97.4318 | 3 |
| 27454785 | Делеция | TAATGAA | TAA | 113.467 | 7 |
| 116423424 | Замена | T | C | 221.999 | 49 |
6. Аннотация SNP.
Работая с программой ANNOVAR, нужно было аннотировать полученные ранее SNP. [5] Для этого из файла была удалена информация об инделях, к новому файлу (chr8_snp_noindl.vcf) я применила скрипт convert2annovar.pl.| Использованная команда | Описание | Результат |
perl /nfs/srv/databases/annovar/convert2annovar.pl | Подготовка файла (перевод в формат ANNOVAR) | chr8_snp.avinput |
Были произведены аннотации SNP по следующим базам данных - refgene, dbsnp, 1000 genomes, GWAS и Clinvar посредством скрипта annotate_variation.pl. В ANNOVAR существуют 3 типа аннотаций, основанных на:
1) gene-based annotation - генной разметке;
2) region-based annotation - разметке других регионов генома;
3) filter-based annotation - фильтрации.
*Для каждой базы указан тип использованной аннотации.
1. Аннотация по базе refgene (gene-based).
| Использованная команда | Описание | Результат |
perl /nfs/srv/databases/annovar/ | Аннотация файлов по генной разметке | chr8_refgene.exonic_variant_function - описание всех полиморфизмов; chr8_refgene.variant_function - описание полиморфизмов в экзонах; chr8_refgene.log - отчет о работе программы |
В первой колонке файла *.variant_annotation отображены категории, указывающие местоположение полиморфизма (в скобках указано число снипов данной категории, встретившихся в моем файле):
- exonic - полиморфизм, попадающий в экзон полностью или частично (5);
- splicing - полиморфизм в пределах n п.о. от границы сплайсинга (n по умолчанию равно 2);
- ncRNA - полиморфизм входит в транскрипт некодирующих РНК;
- UTR5 - полиморфизм входит в 5′-НТО;
- UTR3 - полиморфизм входит в 3′-НТО (13);
- intronic - интронный полиморфизм (58);
- downstream - полиморфизм на расстоянии 1-kb downstream от сайта окончания транскрипции (граница может быть изменена);
- upstream - полиморфизм на расстоянии 1-kb upstream от сайта начала транскрипции (граница может быть изменена);
- intergenic - полиморфизм межгенной области (18).
Таблица экзонных полиморфизмов
| № | Координаты | Ген | Нуклеотидная замена | Тип замены | Качество чтений | Глубина покрытия | АK замена |
| 1 | 27462481 | CLU [6] | A > G (синонимичная) | het | 80.0075 | 16 | H263H |
| 2 | 76452313 | HNF4G [7] | G > A (несинонимичная) | hom | 221.999 | 30 | S29N |
| 3 | 76468228 | HNF4G | G > A (синонимичная) | het | 225.009 | 99 | L209L |
| 4 | 76468282 | HNF4G | G > A (несинонимичная) | hom | 221.999 | 104 | M227I |
| 5 | 116631902 | TRPS1 [8] | C > A (синонимичная) | hom | 221.999 | 35 | P141P, P134P, P132P |
2. Аннотация по базе dbsnp (filter-based).
| Использованная команда | Описание | Результат |
perl /nfs/srv/databases/annovar/ | Аннотация файлов по фильтрации | chr8_snp.hg19_snp138_dropped - полиморфизмы, имеющие идентификатор rs в базе данных dbsnp; chr8_snp.hg19_snp138_filtered - оставшиеся полиморфизмы (не имеют описания rs); chr8_snp.log - отчет о работе программы |
76 полиморфизмов имеют rs-идентификатор, соответственно 18 - не имеют. Интересно отметить, что максимальное покрытие в случае snp без rs составило 5, у большинства этот показатель вообще составляет 1 или 2, в отличие от snp из файла *_dropped.
3. Аннотация по базе 1000 genomes (filter-based).
| Использованная команда | Описание | Результат |
perl /nfs/srv/ | Аннотация файлов по фильтрации | chr8_1000gnm.hg19_ALL.sites.2014_10_dropped - полиморфизмы, имеющие идентификатор rs в базе данных
1000 genomes, кроме этого указана частота; chr8_1000gnm,hg19_ALL.sites.2014_10_filtered - оставшиеся полиморфизмы (не имеют описания rs); chr8_1000gnm.log - отчет о работе программы |
По-прежнему, 76 полиморфизмов имеют указанный идентификатор, частота варьирует от примерно 0,005 до 1.
4. Аннотация по базе Gwas (region-based).
| Использованная команда | Описание | Результат |
perl /nfs/srv/databases/ | Аннотация файлов по разметке регионов генома | chr8_gwas.hg19_gwasCatalog - полиморфизмы, имеющие описанное клиническое значение; chr8_1000gnm.log - отчет о работе программы |
По данным файла можно видеть, что клиническое значение аннотировано в четырёх snp (это болезнь Альцгеймера, регуляция уровня мочевой кислоты, HDL ("хороший") cholesterol), причем среди них нет ни одного экзонного. Следовательно, можно сделать вывод о сложности процессов регуляции и экспрессии генов с данным клиническим значением.
5. Аннотация по базе Clinvar (filter-based).
| Использованная команда | Описание | Результат |
perl /nfs/srv/databases/ | Аннотация файлов по фильтрации | chr8_clinvar.hg19_clinvar_20150629_dropped - полиморфизмы, имеющие аннотацию в базе Clinvar; chr8_clinvar.hg19_clinvar_20150629_filtered - полиморфизмы, не имеющие аннотацию в базе Clinvar; chr8_clinvar.log - отчет о работе программы |
По результатам работы последней программы, не было найдено аннотаций в базе Clinvar (файл *_dropped оказался пустым).