Поиск по сходству (нуклеотидный BLAST).
Задание 1. Определение таксономии и функции прочтенной нуклеотидной последовательности.
В данном практикуме использовалась последовательность WS2966_aligned.fasta, полученная в практикуме 6. По ней был запущен алгоритм blastn (Somewhat similar sequences) с описанными ниже параметрами.
База данных (Database): Nucleotide collection (nr/nt)
Максимально возможное число находок (Max target sequences): 20 000
Порог ожидания (Expect threshold): 10
Длина слова (Word size): 10
Баллы за совпадения и мисматчи (Match/Mismatch scores): 2, -3
Штрафы за гэпы (Gap costs): открытие (Existence) 5; длина (Extension) 2
P.S. Параметры алгоритма заданы по умолчанию
Результаты выдачи blastn
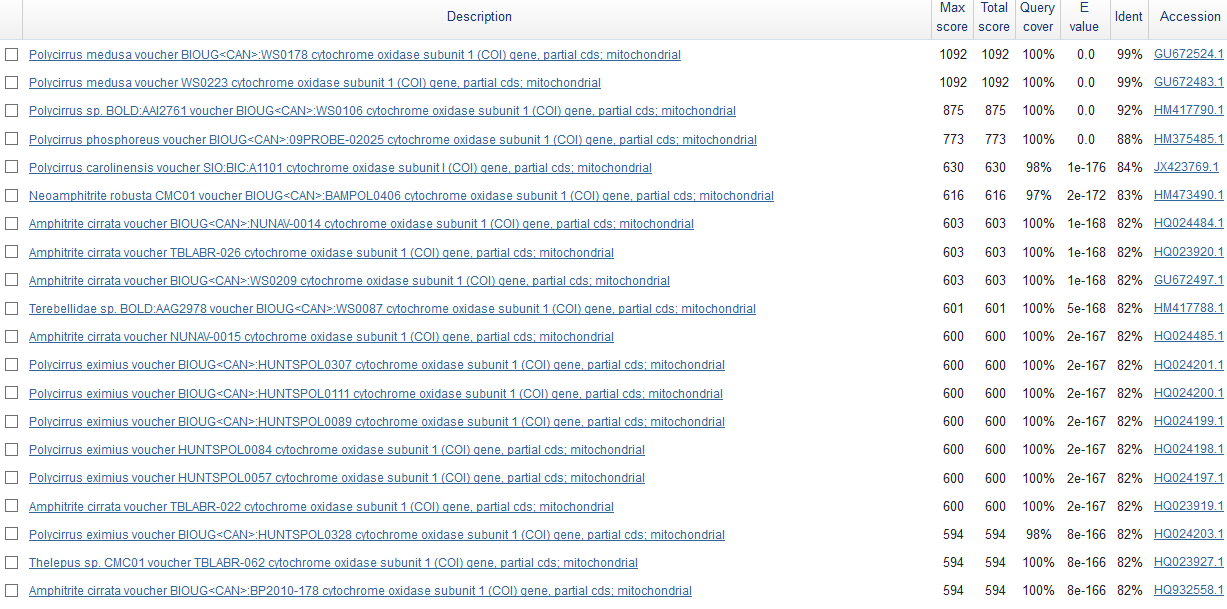
Исходя из полученных результатов, можно сделать вывод, что изучаемая последовательность - митохондриальный ген, кодирующий субъединицу 1 цитохром-С оксидазы дыхательного комплекса IV (терминального каталитического комплекса митохондриального окислительного фосфорилирования). Цитохром С играет ключевую роль в аэробном метаболизме. У человека MT-CO1 (COX1) кодируется геном MT-CO1, а у других эукариот генами COX1, CO1 или COI. [1], [2]
Цитохром-C оксидаза бычьего сердцa
Раскраска по цепям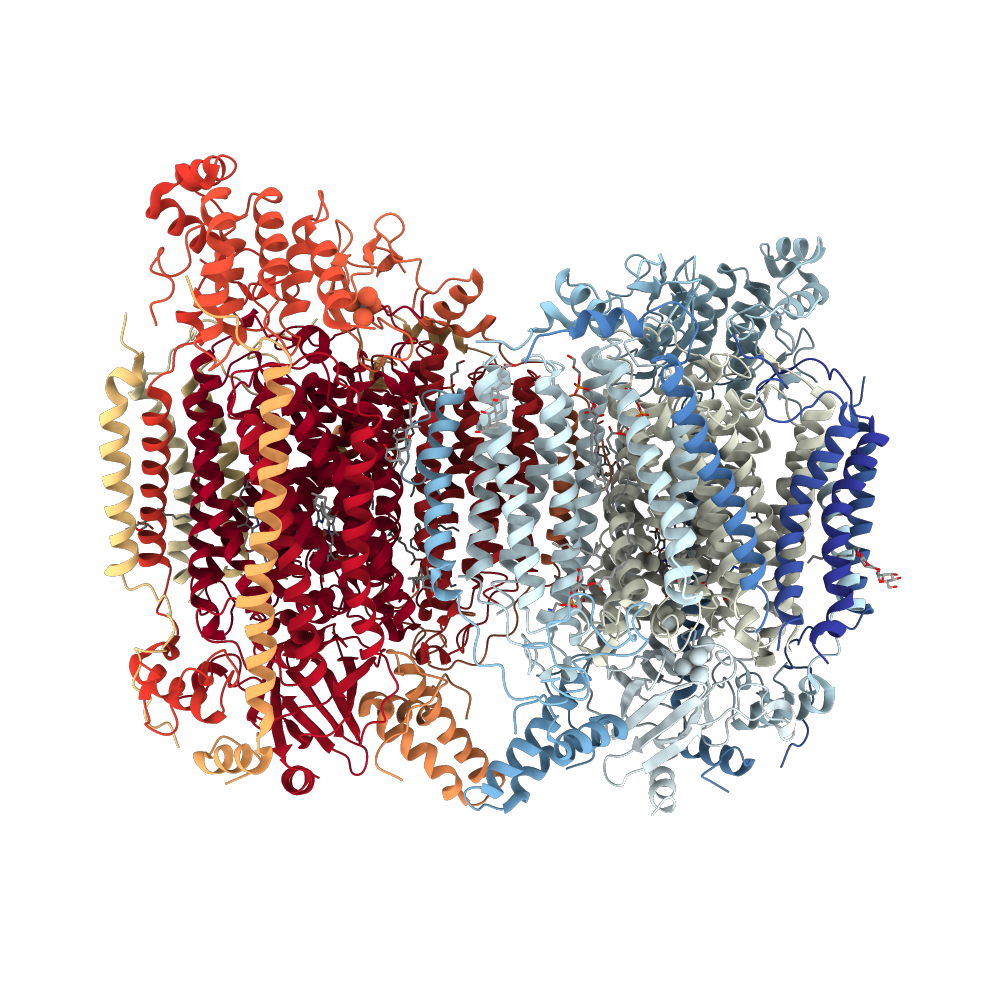 |
Трансмембранный вид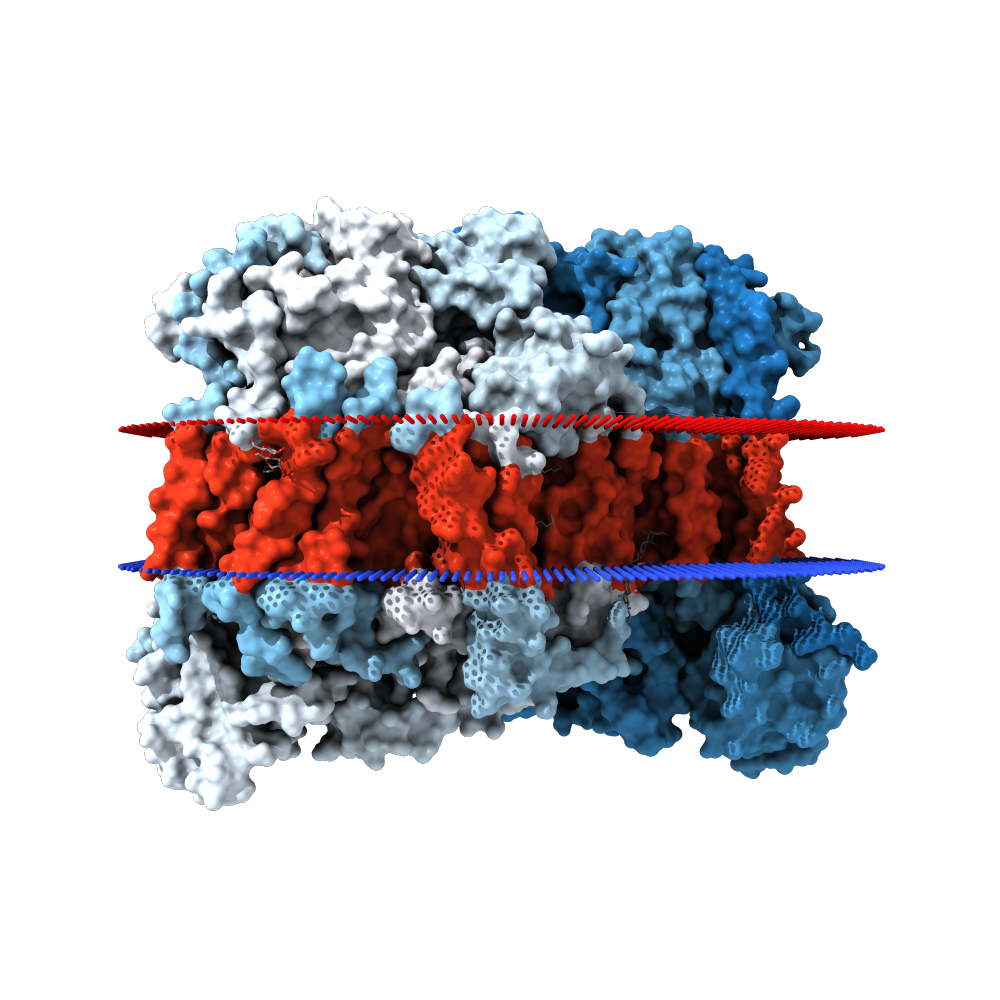 |
Выравнивание последовательностей

После анализа выравнивания, я могу предположить, что данная мне последовательность может принадлежть морскому червю Polycyrrus medusa (так как при сравнении первых двух последовательностей была выявления только одна замена в 474 позиции (C->T)). Количество замен в последовательностях других находок резко отличаются. Например, у Polycymus sp. только на первую сотню нуклеотидов приходится 4 замены, а всего их порядка 40. Вообще говоря, в данных последовательностях достаточно хорошо прослеживается гомология, что подтверждает родственность таксонов, к которым относятся представители, а именно: [3]
Polycirrus medusa [4]
© BIO Photography Group, Biodiversity Institute of Ontario
© BIO Photography Group, Biodiversity Institute of Ontario

Задание 2. Сравнение списков находок нуклеотидной последовательности 3-я разными алгоритмами BLAST.
При помощи параметров запуска (в таблице ниже), был проведен анализ результатов работы трех алгоритмов - blastn (Somewhat similar sequences), discontiguous megablast (More dissimilar sequences) и megablast (Highly similar sequences) с ограничением по таксону Terebellidae.
| Параметры алгоритмов BLAST | |||
|---|---|---|---|
| blastn | discontiguous megablast | megablast | |
| Database | Nucleotide collection (nr/nt) | Nucleotide collection (nr/nt) | Nucleotide collection (nr/nt) |
| Max target sequence | 1000 | 1000 | 1000 |
| Expect threshold | 10 | 10 | 10 |
| Word size | 7 | 11 | 20 |
| Max matches in a query gap | 0 | 0 | 0 |
| Match/Mismatch scores | 2, -3 | 2, -3 | 1, -2 |
| Gap costs | Existence:5 Extension:2 | Existence:5 Extension:2 | Linear |
| Результаты выдачи алгоритмов | ||
|---|---|---|
| blastn | ||
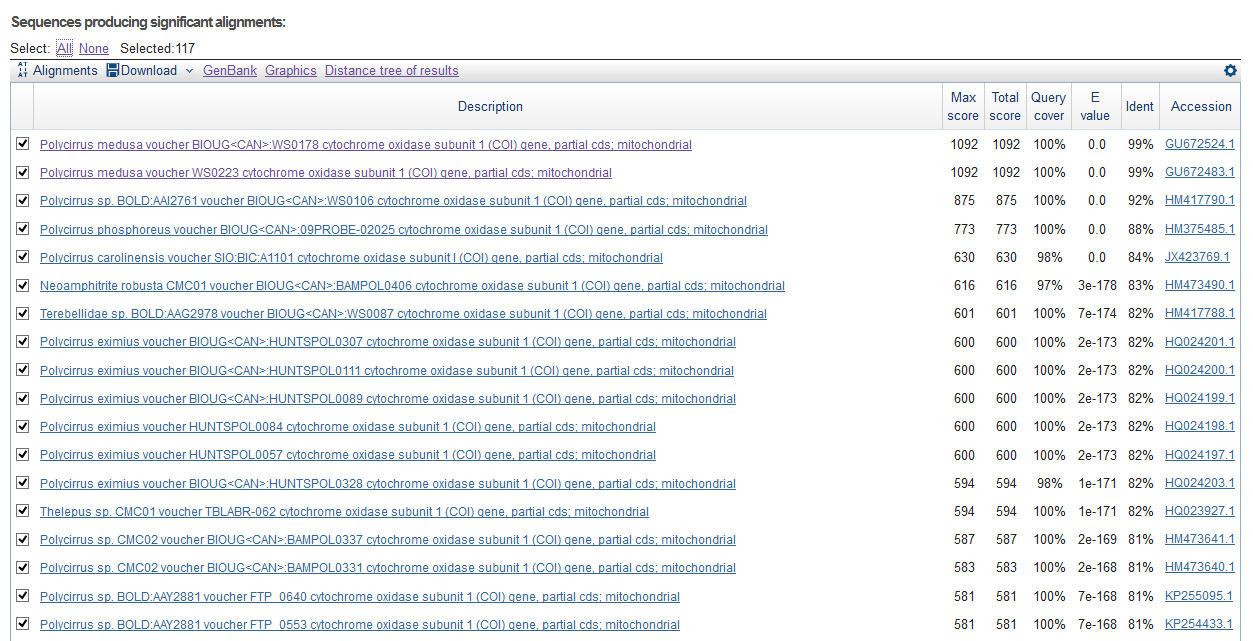 | ||
| discontigous megablast | ||
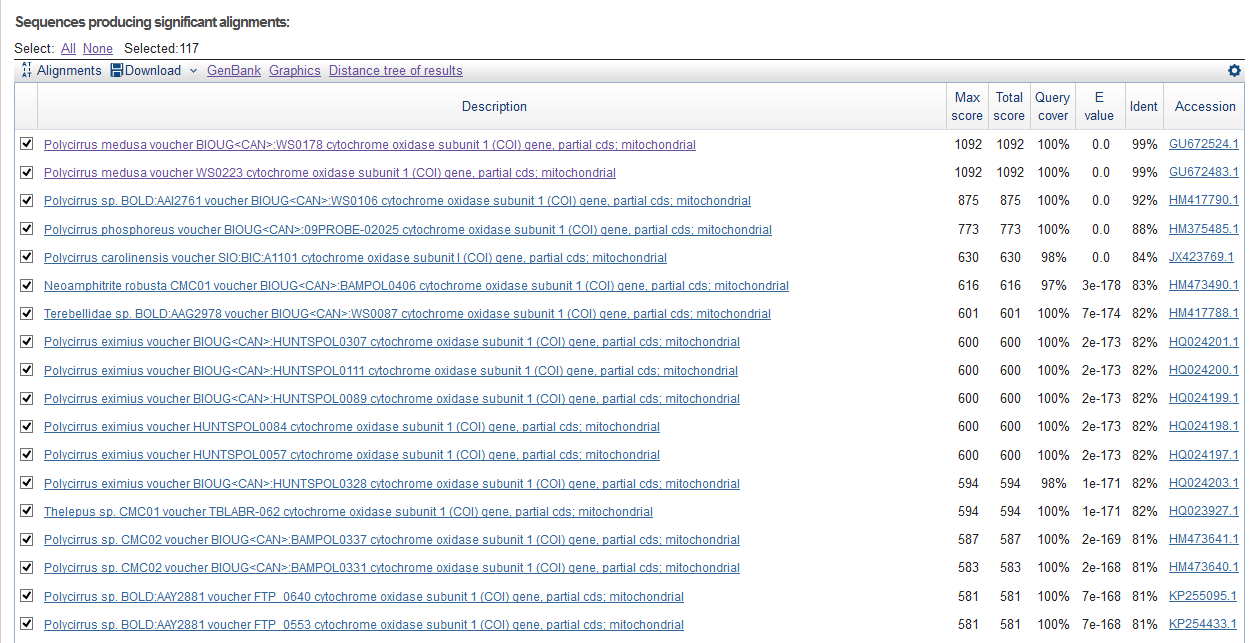 | ||
| megablast | ||
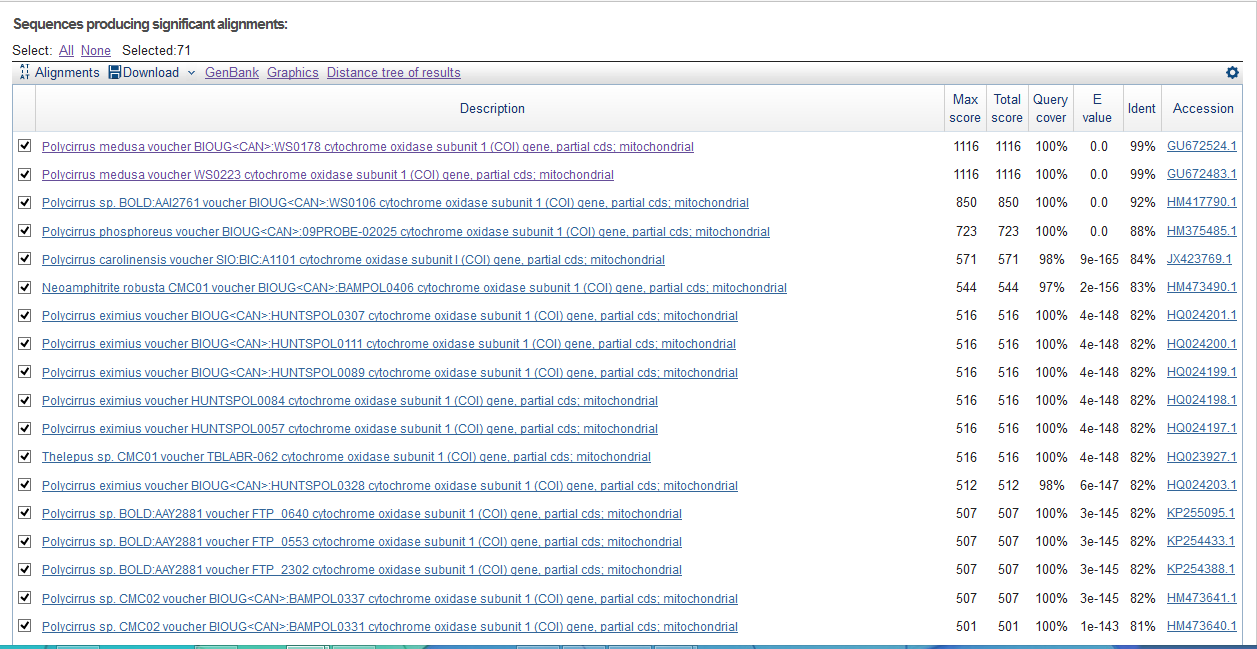 | ||
| Сравнение алгоритмов | |||
|---|---|---|---|
| blastn | discontiguous megablast | megablast | |
| Число находок | 117 | 117 | 72 |
| Max score лучшей находки | 1092 | 1092 | 1116 |
| Max score худшей находки | 203 | 203 | 248 |
| E-value лучшей находки | 0.0 | 0.0 | 0.0 |
| E-value худшей находки | 7e-54 | 7e-54 | 2e-67 |
| Identity лучшей находки (%) | 99 | 99 | 99 |
| Identity худшей находки (%) | 67 | 67 | 75 |
| Лучшее Query cover | 100 | 100 | 100 |
| Худшее Query cover | 75 | 75 | 75 |
P.S. Из таблицы были удалены находки со значениями E-value >0.001 и Query cover <60%, так как они не учитывались при анализе.
Как можно видеть из данных таблицы, результаты работы blastn и discontiguous megablast по случайному совпадению оказались идентичными, поэтому адекватную оценку при сравнении данных алгоритмов получить было сложно. Зато очевидна их разница в работе относительно megablast. 41 находка, которая была найдена с помощью blastn и discontiguous megablast не найдена в megablast. Соответственно все находки из выдачи megablast, были найдены в каждом случае.
Первые 5 находок (самые лучшие) одинаковы во всех алгоритмах. В выдаче blastn и discontiguous megablast были негомологичные находки, не подходящие по рангу E-value, Query или Identity cover, что говорит о меньшей точности алгоритмов. К примеру, E-value худшей находки blastn составляет 7e-54, в то время как у megablast 2e-67. Но также megablast отсеивает и "подозрительно хорошие" находки, оставляя, вероятно, наиболее гомологичные, что возможно благодаря большой длине Word size, которая по умолчанию составляет 28 (я выбрала 20).
Пример находки, которая была обнаружена только blastn и discontiguous blast: Terebellidae sp. BOLD:AAG2978 voucher BIOUG:WS0087 cytochrome oxidase subunit 1 (COI) gene, partial cds; mitochondria. Ee Max score=Total score - 601; Query cover - 100,00%; E-value - 7,00E-174; Ident - 82,00%. На схеме отсутствуют участки длиной 20, что и объясняет отсутствие соответствующей ей последовательности в результатах выдачи megablst.
Sequence ID: HM417788.1 Length: 660 Number of Matches: 1 Range 1: 29 to 639
Score Expect Identities Gaps Strand
601 bits(666) 7e-174 499/611(82%) 0/611(0%) Plus/Plus
Query 1 GAGGAGGATTATTAGGAACCTCTATAAGACTACTCATCCGAATTGAACTTGGCCAACCTG 60
||||||| | | || || ||||||||||| || || ||| | ||| | |||||||| |
Sbjct 29 GAGGAGGCCTTCTTGGTACTTCTATAAGACTTCTAATTCGAGTAGAATTAGGCCAACCAG 88
Query 61 GTGCTTTTCTTGGAAGAGACCAATTATATAACACTATTGTAACTGCTCACGGGCTACTTA 120
| || || | || |||||||||||||| || |||||||| || || ||||| ||||| |
Sbjct 89 GAGCCTTCTTAGGCAGAGACCAATTATACAATACTATTGTTACCGCCCACGGCCTACTAA 148
Query 121 TAATCTTTTTTCTTGTTATACCTGTACTTATTGGAGGTTTTGGAAACTGATTAATCCCAC 180
|||| || ||| | || |||||||| | || || || || |||||||||||||||||
Sbjct 149 TAATTTTCTTTTTAGTAATACCTGTTTTAATCGGGGGGTTCGGAAACTGATTAATCCCTT 208
Query 181 TTATATTAGGCGCACCAGACATAGCCTTCCCACGAATAAACAATATAAGGTTTTGACTAC 240
| |||||||| |||||||| ||||| ||||| || ||||||||| |||| ||||| ||||
Sbjct 209 TAATATTAGGAGCACCAGATATAGCTTTCCCGCGTATAAACAATTTAAGATTTTGGCTAC 268
Query 241 TCCCTCCTGCACTACTTYTYCTCCTCGCTTCCGCAGCAGTTGAAAAAGGAGTAGGAACTG 300
|||| ||||| || ||| | || || || |||||||| ||||||||||| || || |
Sbjct 269 TCCCGCCTGCTCTTCTTCTTCTTCTTAGCTCTGCAGCAGTAGAAAAAGGAGTTGGGACAG 328
Query 301 GTTGGACTCTTTACCCCCCTTTAGCAAGAAATCTAGCACATGCTGGACCCTCAGTAGACC 360
|||||||| ||||||| |||||||| |||||| |||| |||||||| || ||||||||||
Sbjct 329 GTTGGACTGTTTACCCTCCTTTAGCTAGAAATATAGCCCATGCTGGCCCATCAGTAGACC 388
Query 361 TTGCTATTTTTTCACTACATTTAGCTGGTATTTCCTCAATTTTAGGAGCTATTAACTTTA 420
|||| |||||||| |||||||||| ||||| || |||||| | ||||| ||||| ||||
Sbjct 389 TTGCAATTTTTTCTTTACATTTAGCAGGTATCTCTTCAATTCTTGGAGCCATTAATTTTA 448
Query 421 TCACAACAGTAGCAAATATACGATGAAAAGGACTACGTCTAGAACGAGTTCCTCTATTTG 480
| ||||| || || |||||||||||||||||| ||||||| || ||| ||||| ||||||
Sbjct 449 TTACAACTGTTGCTAATATACGATGAAAAGGATTACGTCTTGAGCGAATTCCTTTATTTG 508
Query 481 TTTGAGCTGTAGATATCACTGTTGTTCTACTACTTTTATCCCTCCCTGTATTAGCCGGAG 540
|||||||||| ||||||| ||| |||| || ||||||||| | || || ||||| || |
Sbjct 509 TTTGAGCTGTTAATATCACAGTTATTCTCCTTCTTTTATCCTTACCCGTTTTAGCAGGGG 568
Query 541 CAATTACTATATTATTAACAGACCGAAATGTTAACACATCATTCTTTGACCCTAGAGGAG 600
|||| || ||| | ||||||||||| ||||||||||| ||||| |||||||| |||||||
Sbjct 569 CAATCACAATACTTTTAACAGACCGTAATGTTAACACCTCATTTTTTGACCCAAGAGGAG 628
Query 601 GAGGAGACCCA 611
|||||||||||
Sbjct 629 GAGGAGACCCA 639Выводы:
1) blastn выдает достаточно большое количество последовательностей (достаточно коротких), однако далеко не все из них могут являться гомологами изучаемой, удобно использовать при идентификации конкретной нуклеотидной последовательности, сравнивания с уже имеющимися последовательностями в базах данных;
2) discontiguous megablast может быть использован при изучении дивергировавших гомологов во время межвидового анализа;
3) megablast удобен при внутривидовом анализе, подходит для выравнивания длинных последовательностей.
Задание 3. Проверка наличия гомологов трех белков у Amoboaphelidium protococarum.
Благодаря использованию локального BLAST в этом задании были проверены на наличие гомологов в геноме организма X5 (Amoeboaphelidium protococarum) следующие белки: HSP7C_HUMAN, CISY_HUMAN, RPB2_HUMAN.
Указанный организм относится к афелидам, родственному настоящим грибам таксону. Все описанные виды - внутриклеточные паразиты водорослей с фаготрофной амебоидной вегетативной стадией, инвазивная циста с короткой инфекционной трубкой аппарата проникновения, зооспоры с псевдоподиями и/или направленным назад функционирующим (возможно, рудиментарным) жгутиком. [5]
Amoeboaphelidium protococarum
Сначала я создала локальную базу данных генома Amoeboaphelidium protococarum:
makeblastdb -in X5.fasta -dbtype nucl
После этого для каждого белка был запущен алгоритм tblastn, находящий гомологи белка в трансляции нуклеотидного банка (т.е. данного нам генома):
tblastn -query xxx.fasta -db X5.fasta -out xxx.out -outfmt 7
| Описание результатов выдачи алгоритма | |
|---|---|
HSP7C_HUMAN | |
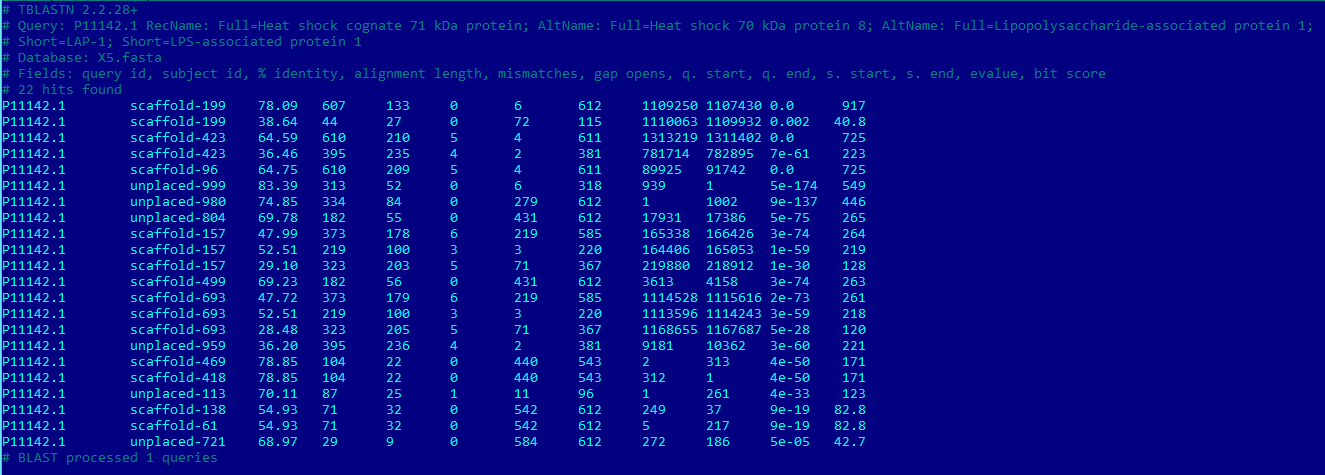 | |
Белок HSP7C - белок теплового шока. Выступает репрессором активации транскрипции.
Ингибирует транскрипционную активность CITED1 Smad-опосредованной транскрипции. Консервативный шаперон HSP70. Компонент комплекса PRP19-CDC5L, формирующий концевую часть
сплайсосомы, необходим для активации сплайсинга pre-mRNA. Контактирует со всеми ее компонентами, поэтому может выполнять структурную функцию. Связывается с бактериальными
LPS, выступает посредником в LPS-индуцированной воспалительной реакции, включая секрецию моноцитами фактора некроза опухолей TNF. [7] Белки Hsp70 имеют 3 главных функциональных домена. На N-конце — АТФ-связывающий домен, гидролизует ее до АДФ. Между N- и С- концами находится консервативный домен (петля), связывающий субстрат. Домен на C-концевом участке с выраженной альфа-спиральной структурой выступает в качестве лида для субстрат-связывающего домена. [8] По результатам алгоритма для данного белка было получено 22 находки. Лучшая из них (scaffold-199) с большой вероятностью гомологична. Ее параметры таковы: > scaffold-199 Length=1112851 Score = 917 bits (2369), Expect = 0.0, Method: Compositional matrix adjust. Identities = 474/607 (78%), Positives = 538/607 (89%), Gaps = 0/607 (0%) Frame = -2Кроме того, среди полученных выравниваний встречались и те, куда вошли определенные участки изучаемого белка, что говорит о гомологии доменов. В качестве примера можно рассмотреть unplaced-999, возможно, являющегося частью более крупного домена-гомолога (его точное расположение не определено): > unplaced-999 Length=6853 Score = 549 bits (1414), Expect = 5e-174, Method: Compositional matrix adjust. Identities = 261/313 (83%), Positives = 288/313 (92%), Gaps = 0/313 (0%) Frame = -2По данным Uniprot участку с 1 по 386 позицию на N-терминальном конце соответствует домен АТФазы, следовательно, в протеоме Amoeboaphelidium protococarum есть гомологичный данному домену. | |
| Выдача tblastn по HSP7C Выдача tblastn по HSP7C c комментариями | |
CISY_HUMAN | |
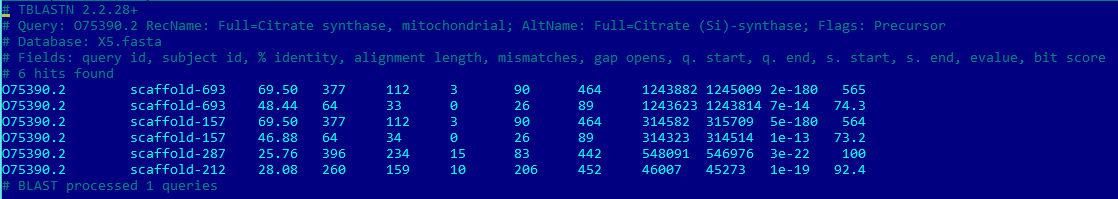 | |
Белок CISY_HUMAN - митохондриальная цитрат-синтаза. Проявляет каталитическую
активность в реакции: Acetyl-CoA + H2O + oxaloacetate = citrate + CoA. Участвует в цикле Кребса (трикарбоновых кислот). Располагается в митохондриальном матриксе эукариот,
но закодирован в ядре. Синтезируется на цитоплазматических рибосомах, затем транспортируется в митохондрии. [9], [10] Из 6 полученных находок (в двух скэффолдах их получилось по 2) лучшей оказалась scaffold-693: > scaffold-693 Length=1268102 Score = 565 bits (1457), Expect = 2e-180, Method: Compositional matrix adjust. Identities = 262/377 (69%), Positives = 315/377 (84%), Gaps = 3/377 (1%) Frame = +1Следует отметить ее фактическую идентичность со scaffold-157 (с разницей 1 в BitScore, E-value немного больше — 5e-180 сравнительно с 2e-180 у scaffold-693). За исключением различий рамки считвыания выравнивания из этих двух скэффолодов с последовательностью CISY_HUMAN были практически одинаковым. Параметры выравниваний говорят о его хорошем качестве, а также о гомологии исследуемого белка без изменения функций. | |
Выдача tblastn по CISY Выдача tblastn по CISY c комментариями | |
RPB2_HUMAN | |
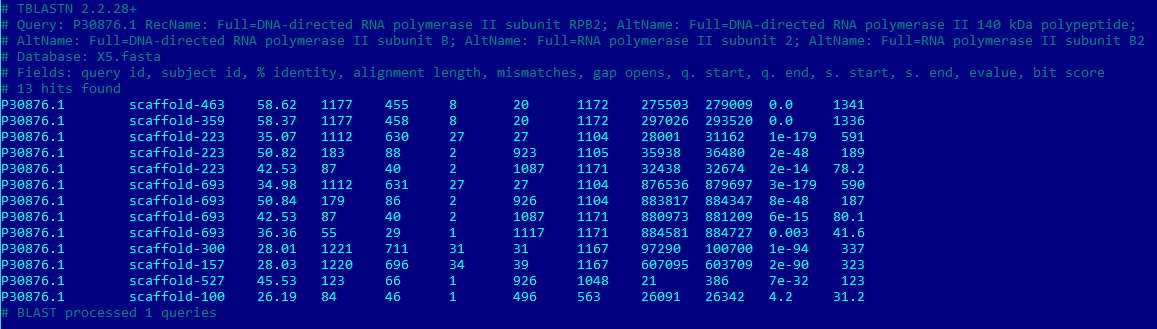 | |
Белок RPB2_HUMAN - ДНК-зависимая РНК-полимераза, катализирует транскрипцию
ДНК в РНК с использованием в качестве субстрата рибонуклеозидтрифосфаты. Синтезирует предшественники мРНК и множество некодирующих РНК. [11] Фермент, который кодируется геном POLR2B у человека. Данный
ген кодирует вторую по величине субъединицу РНК-полимеразы II, ответственной за синтез РНК у эукариот. [12] Из 13 находок параметры первых двух очень схожи: > scaffold-463 Length=442349 Score = 1341 bits (3470), Expect = 0.0, Method: Compositional matrix adjust. Identities = 690/1177 (59%), Positives = 879/1177 (75%), Gaps = 32/1177 (3%) Frame = +1Остальные же, на мой взгляд, вряд ли можно назвать гомологичными белку исследуемого организма ввиду слишком низких параметров. Качество покрытия двух лучших выравниваний позволяет говорить об условно положительной гомологии (вероятно, только один из доменов консервативен, остальные же у человека и Amoeboaphelidium protococarum могут различаться). | |
Выдача tblastn по RPB2 Выдача tblastn по RPB2 c комментариями | |
Задание 4. Поиск гена белка, закодированного в одном скэффолде Amoeboaphelidium.
Для выполнения задания в геноме Amoeboaphelidium protococarum был выбран scaffold-17. С помощью следующей команды я получила информацию о длине скэффолдов:
infoseq X5.fasta -only -name -length
seqret X5.fasta:scaffold-17 -out scaf17.fasta
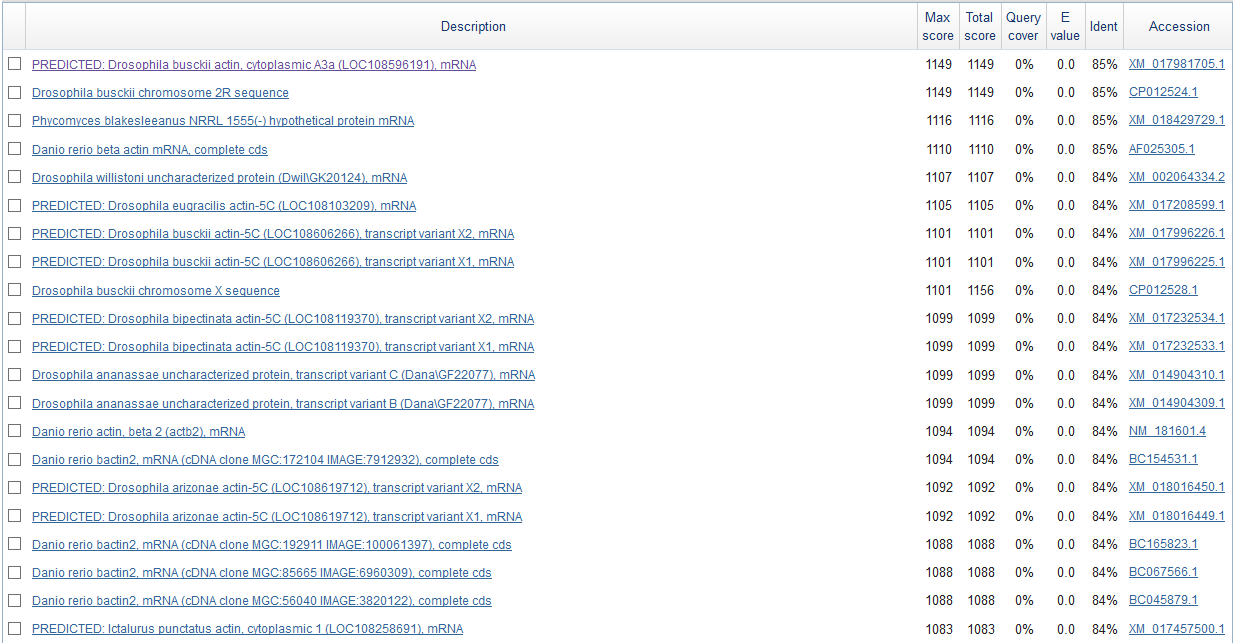
Выдача megablast с ограничением Opisthokonta
Выдача megablast с ограничением Fungi
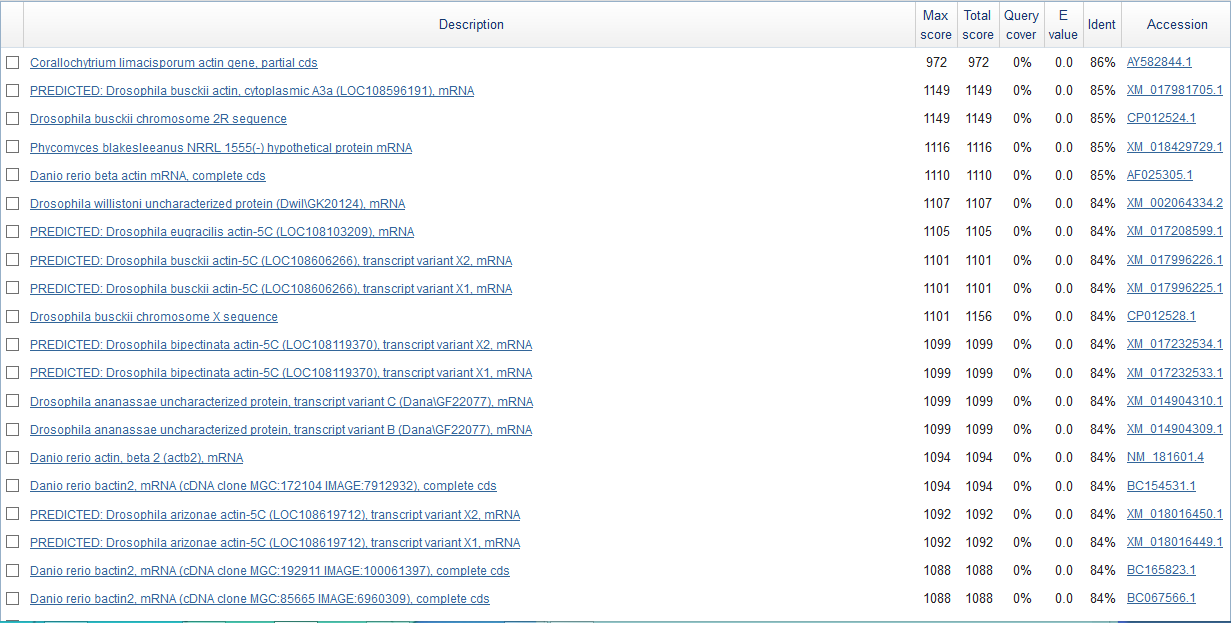
Как можно видеть, процент идентичности последовательностей достаточно высок, а E-value равен 0.0. Нулевой процент покрытия объясняется малой длиной гена в сравнении с двухмиллионным скэффолдом. Исходя из результатов работы алгоритма, предполагаю, что в scaffold-17 закодирован ген актина.
Источники
- [1] Wikipedia: Cytochrome C Oxidase Subunit I
- [2] PDB: Bovine heart cytochrome C oxidase
- [3] WoRMS taxon tree
- [4] EOL: Polycirrus medusa
- [5] Peter M. Letcher, Martha J. Powell. A new isolate of Amoeboaphelidium protococcarum, and Amoeboaphelidium occidentale, a new species in phylum Aphelida (Opisthosporidia). Mycologia: 107 (3), 522-531. 2015 Feb 06.
- [6] Kodomo: BLAST
- [7] Uniprot: Heat shock cognate 71 kDa protein
- [8] Wikipedia: Hsp70
- [9] Uniprot: Citrate synthase, mitochondrial
- [10] Wikipedia: Citrate synthase
- [11] Uniprot: DNA-directed RNA polymerase II subunit RPB2
- [12] Wikipedia: POLR2B