Evolutionary trees.
Задание 1. Построение дерева по нуклеотидным последовательностям.
Для выполнения задания мной были получены последовательности 16S рибосомальной РНК каждой из бактерий (указанных в
первом практикуме данного блока) из базы полных геномов NCBI (файлы с расширением .frn
с РНК-кодирующими участками генома). Штаммы бактерий и последовательности для выравнивания выбирались случайным образом.
Из полного генома бактерий для каждой было отобрано по одной последовательности, кодирующей субъединицу 16S (во многих геномах таких последовательностей несколько). Выбранные последовательности были объединены в fasta-файл (с указанием в качестве идентификаторов только мнемоник видов) для дальнейшего выравнивания в JalView алгоритмом Muscle. По данному выравниваю в MEGA было построено дерево.
Из полного генома бактерий для каждой было отобрано по одной последовательности, кодирующей субъединицу 16S (во многих геномах таких последовательностей несколько). Выбранные последовательности были объединены в fasta-файл (с указанием в качестве идентификаторов только мнемоник видов) для дальнейшего выравнивания в JalView алгоритмом Muscle. По данному выравниваю в MEGA было построено дерево.
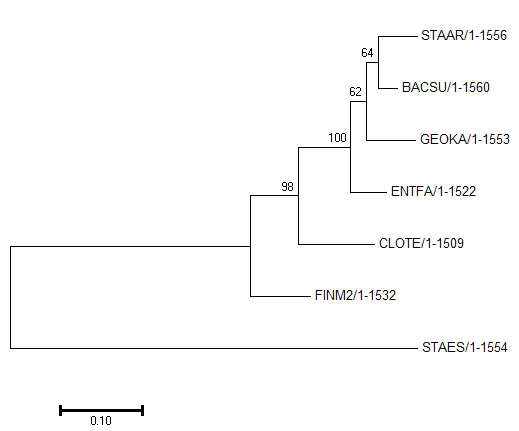
Дерево, построенное на основе выравнивания последовательностей 16S RNA методом NJ
NJ + Bootstrap-проверка: Original tree
NJ + Bootstrap-проверка: Consensus tree
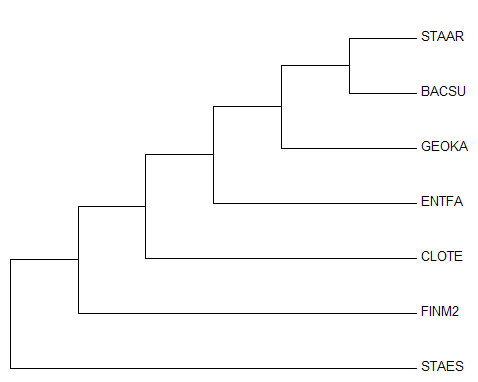
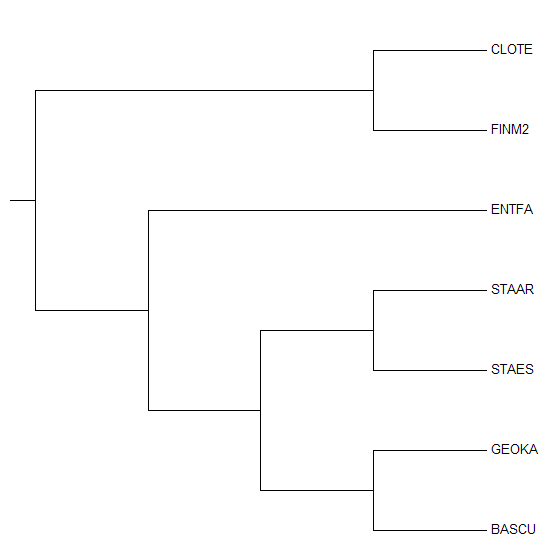
Эталонное дерево
Топология дерева, построенного по рибосомальной РНК:
1) {STAES, FINM2} vs. {CLOTE, ENTFA, GEOKA, BACSU, STAAR}
2) {STAES, FINM2, CLOTE} vs. {ENTFA, GEOKA, BACSU, STAAR}
3) {STAES, FINM2, CLOTE, ENTFA, GEOKA} vs. {BACSU, STAAR}
Мы видим, что в данном дереве нет ни одной нетривиальной ветви, совпадающей с ветвями эталонного дерева. Также можно наблюдать неправильное укоренение в ветвь {STAES} vs. {FINM2, CLOTE, ENTFA, GEOKA, BACSU, STAAR}. Такие результаты вполне объяснимы, ведь метод NJ относится к дистаницонным, то есть оценивает расстояния между последовательностями, учитывая число мутаций, а судя по выравниванию, последовательность STAES явно выделяется большим количеством инделей.
1) {STAES, FINM2} vs. {CLOTE, ENTFA, GEOKA, BACSU, STAAR}
2) {STAES, FINM2, CLOTE} vs. {ENTFA, GEOKA, BACSU, STAAR}
3) {STAES, FINM2, CLOTE, ENTFA, GEOKA} vs. {BACSU, STAAR}
Мы видим, что в данном дереве нет ни одной нетривиальной ветви, совпадающей с ветвями эталонного дерева. Также можно наблюдать неправильное укоренение в ветвь {STAES} vs. {FINM2, CLOTE, ENTFA, GEOKA, BACSU, STAAR}. Такие результаты вполне объяснимы, ведь метод NJ относится к дистаницонным, то есть оценивает расстояния между последовательностями, учитывая число мутаций, а судя по выравниванию, последовательность STAES явно выделяется большим количеством инделей.
Выравнивание последовательностей 16S RNA

Если говорить о сходствах с деревьями, реконструированными по белковым последовательностям, можно лишь отметить общую тенденцию соседнего
расположения листьев CLOTE и FINM2 (в эталонном дереве они объединены в кладу) и отделение группы (STAAR, BACSU, GEOKA) и Enterococcus faecalis от общего
предка.
*Попытка что-то исправить*
Из-за плохого выравнивания STAES относительно других последовательностей была предпринята попытка обновить сами последовательности (выбирались близкие по длине, с максимальным различием в 15 нуклеотидов). Однако и такой подход не позволил добиться хорошего качества выравнивания относительно STAES. Поэтому было решено удалить эту последовательность из выравнивания и построить дерево без нее (будем считать, что веть с листом STAAR на самом деле включает в себя 2 листа - STAAR и STAES, как это было в исходном дереве).
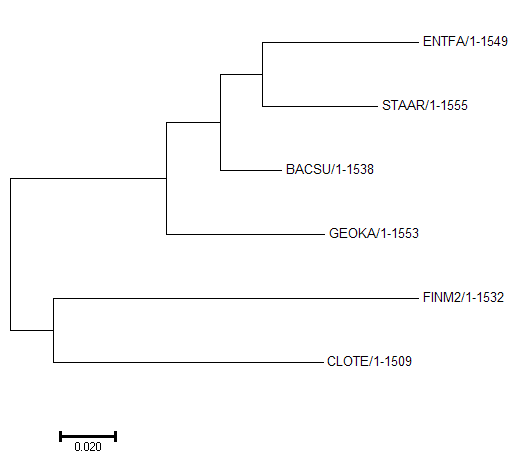
В таком случае наблюдается одна нетривиальная ветвь из эталонного дерева, а именно: {CLOTE, FINM2} vs. {ENTFA, STAAR (+STAES), GEOKA, BASCU}. Результат меня удивил, потому что качество выравнивания без STAES показалось мне вполне удовлетворительным.
*Попытка что-то исправить*
Из-за плохого выравнивания STAES относительно других последовательностей была предпринята попытка обновить сами последовательности (выбирались близкие по длине, с максимальным различием в 15 нуклеотидов). Однако и такой подход не позволил добиться хорошего качества выравнивания относительно STAES. Поэтому было решено удалить эту последовательность из выравнивания и построить дерево без нее (будем считать, что веть с листом STAAR на самом деле включает в себя 2 листа - STAAR и STAES, как это было в исходном дереве).
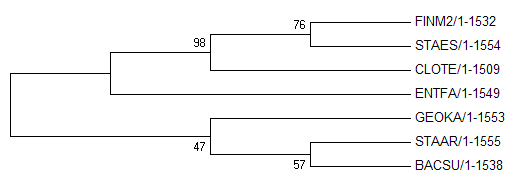
Дерево, построенное на основе выравнивания последовательностей 16S RNA (без STAES) методом NJ
В таком случае наблюдается одна нетривиальная ветвь из эталонного дерева, а именно: {CLOTE, FINM2} vs. {ENTFA, STAAR (+STAES), GEOKA, BASCU}. Результат меня удивил, потому что качество выравнивания без STAES показалось мне вполне удовлетворительным.
Задание 2. Построение и анализ дерева, содержащего паралоги.
В данном задании предлагалось построить дерево гомологичных белку CLPX_BACSU белков
заданного списка бактерий. Затем, полагая, что дерево реконструировано верно, нужно было определить ортологи и паралоги, привести примеры некоторых эволюционных
событий.
Файлы из директории P:\y15\term4\Proteomes, содержащие полные протеомы (база UniProt), были конкатенированы в файл proteoms.fasta.
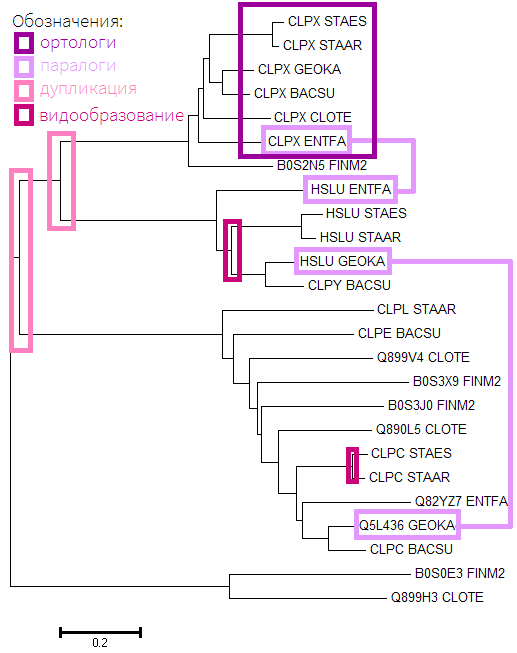
| Ортологи - пара гомологичных белков из разных организмов, разделение общего предка которых на линии,
ведущие к этим белкам, произошло в результате видообразования. Паралоги - пара гомологичных белков из одного организма. |
Файлы из директории P:\y15\term4\Proteomes, содержащие полные протеомы (база UniProt), были конкатенированы в файл proteoms.fasta.
cat file1 >> file2Далее на основе протеомов была создана база данных, по которой производился поиск гомологов.
makeblastdb -in proteoms.fasta -dbtype prot -out dbprot.fasta blastp -query CLPX_BACSU.fasta -evalue 0.001 -db dbprot.fasta -out resultТаким образом нашлась 31 последовательность, из них 26 достоверных гомологов (с E-value < 1e-4).
Последовательности гомологов
| Последовательность и мнемоника | Bit-Score | E-value |
| sp|P50866|CLPX_BACSU ATP-dependent Clp protease ATP-binding subunit ClpX | 841 | 0.0 |
| sp|Q5KWJ9|CLPX_GEOKA ATP-dependent Clp protease ATP-binding subunit ClpX | 711 | 0.0 |
| sp|Q8CNY5|CLPX_STAES ATP-dependent Clp protease ATP-binding subunit ClpX | 608 | 0.0 |
| sp|Q6GG31|CLPX_STAAR ATP-dependent Clp protease ATP-binding subunit ClpX | 606 | 0.0 |
| sp|Q891J8|CLPX_CLOTE ATP-dependent Clp protease ATP-binding subunit ClpX | 595 | 0.0 |
| sp|Q833M7|CLPX_ENTFA ATP-dependent Clp protease ATP-binding subunit ClpX | 579 | 0.0 |
| tr|B0S2N5|B0S2N5_FINM2 ATP-dependent Clp protease ATP-binding subunit ClpX | 559 | 0.0 |
| sp|Q5L0N1|HSLU_GEOKA ATP-dependent protease ATPase subunit HslU | 101 | 3e-23 |
| sp|Q834K4|HSLU_ENTFA ATP-dependent protease ATPase subunit HslU | 101 | 3e-23 |
| sp|P39778|CLPY_BACSU ATP-dependent protease ATPase subunit ClpY | 99.8 | 1e-22 |
| sp|Q8CPH0|HSLU_STAES ATP-dependent protease ATPase subunit HslU | 99.0 | 2e-22 |
| sp|Q6GHI1|HSLU_STAAR ATP-dependent protease ATPase subunit HslU | 97.4 | 6e-22 |
| tr|Q5L436|Q5L436_GEOKA ATP-dependent Clp protease ATPase subunit | 65.9 | 2e-11 |
| tr|Q890L5|Q890L5_CLOTE Negative regulator of genetic competence ClpC/MecB | 57.4 | 6e-09 |
| sp|P37571|CLPC_BACSU Negative regulator of genetic competence ClpC/MecB | 56.6 | 9e-09 |
| sp|O31673|CLPE_BACSU ATP-dependent Clp protease ATP-binding subunit ClpC | 55.5 | 2e-08 |
| sp|Q6GDQ0|CLPL_STAAR ATP-dependent Clp protease ATP-binding subunit ClpL | 54.7 | 4e-08 |
| sp|Q6GJE4|CLPC_STAAR ATP-dependent Clp protease ATP-binding subunit ClpC | 54.3 | 6e-08 |
| tr|Q899V4|Q899V4_CLOTE Negative regulator of genetic competence ClpC/MecB | 53.5 | 9e-08 |
| sp|Q8CQ88|CLPC_STAES ATP-dependent Clp protease ATP-binding subunit ClpC | 52.0 | 3e-07 |
| tr|B0S3X9|B0S3X9_FINM2 ATP-dependent protease Clp ATP-binding subunit | 51.6 | 4e-07 |
| tr|B0S0E3|B0S0E3_FINM2 ATP-dependent zinc metalloprotease FtsH | 50.1 | 1e-06 |
| tr|B0S3J0|B0S3J0_FINM2 Chaperone protein ClpB OS=Finegoldia magna | 45.4 | 3e-05 |
| tr|Q82YZ7|Q82YZ7_ENTFA ATP-dependent Clp protease, ATP-binding subunit | 45.4 | 3e-05 |
| tr|Q899H3|Q899H3_CLOTE ATP-dependent zinc metalloprotease FtsH | 44.7 | 5e-05 |
По мнемоникам файла result с использованием сервера MUSCLE было
построено выравнивание, на основе которого в MEGA методом Neighbour Joining было реконструировано дерево.
Дерево гомологов белка CLPX BACSU, полученное в MEGA (с учетом длин ветвей, не отображены с эстетической точки зрения)
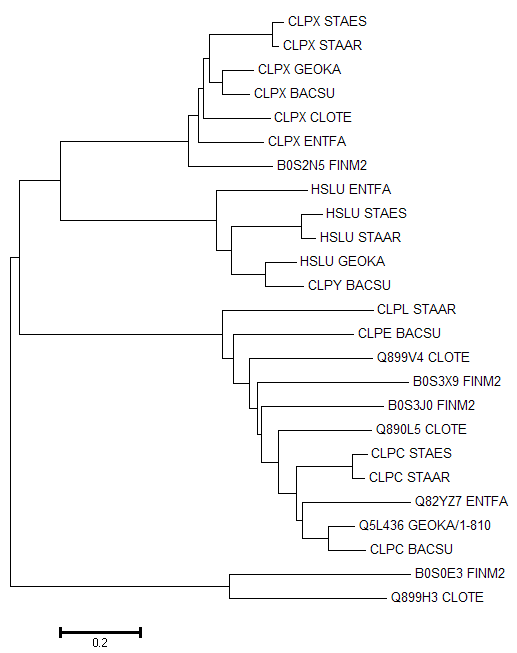
То же дерево с указанием групп гомологов и эволюционных событий