Сначала напишем SMILES для NAG.nag.smi
Сделали pdb.nag.pdb
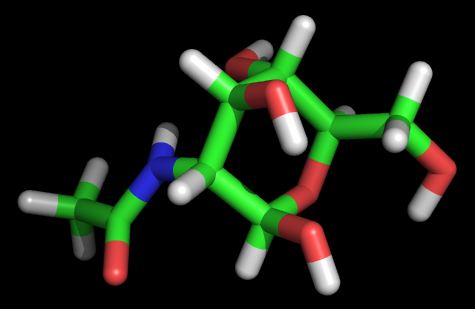
Скриптом prepare_ligand4.py из пакета Autodock tools создаем pdbqt файл лиганда.nag.pdbqt
Скриптом prepare_receptor4.py из пакета Autodock tools создан pdbqt файл белка.seq5.pdbqt
Запустим докинг:
vina --config vina.cfg --receptor seq5.pdbqt --ligand nag.pdbqt --out nag_prot.pdbqt --log nag_prot.logПолучили 2 файла:
nag_prot.log
nag_prot.pdbqt
Просмотрим файл nag_prot.log и запишем энергии 3 лучших расположений и геометрическую разницу между ними:
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -6.4 0.000 0.000
2 -6.2 1.280 2.702
3 -5.7 5.498 7.818
Файлы nag_prot.pdbqt и seq.pdbqt были загружены в PyMOL.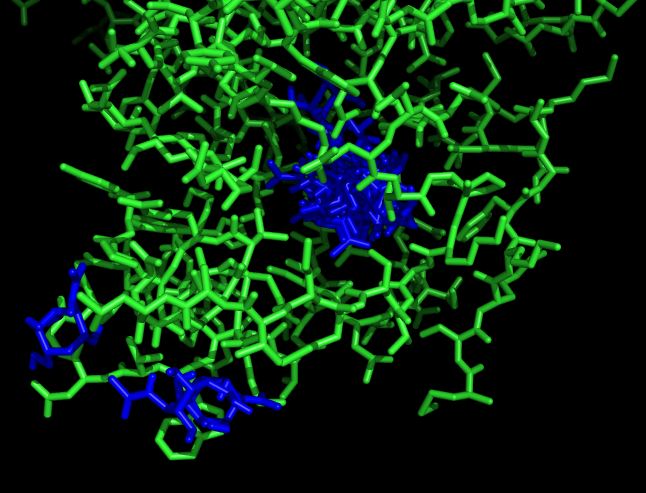

Теперь проведём докинг рассматривая подвижность некоторых боковых радикалов белка.
python /usr/share/pyshared/AutoDockTools/Utilities24/prepare_flexreceptor4.py -r seq5.pdbqt -s ASN45_ASP59_ALA139 vina --config vina.cfg --receptor seq02_rigid.pdbqt --flex seq5_flex.pdbqt --ligand nag.pdbqt --log nag_prot_flex.log --out nag_seq5_flex.pdbqtТак получили 2 файла:
nag_prot_flex.log
nag_seq5_flex.pdbqt
Просмотрим файл nag_prot.log и запишем энергии 3 лучших расположений и геометрическую разницу между ними:
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.8 0.000 0.000
2 -5.8 1.730 2.306
3 -5.7 2.119 2.959
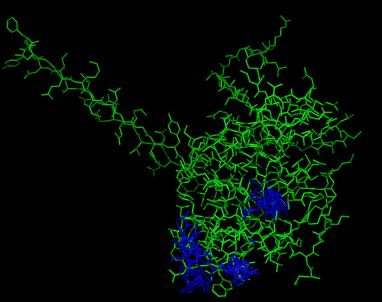

Видно что такой лучше обычного докинга, т.к двигается не только лиганд но аминокслоты,которые мы задали. Так как белок не жесткая струтура, то лучше использовать такой докинг.