Практикум 12. Анализ транскриптомов. Bedtools
Задание
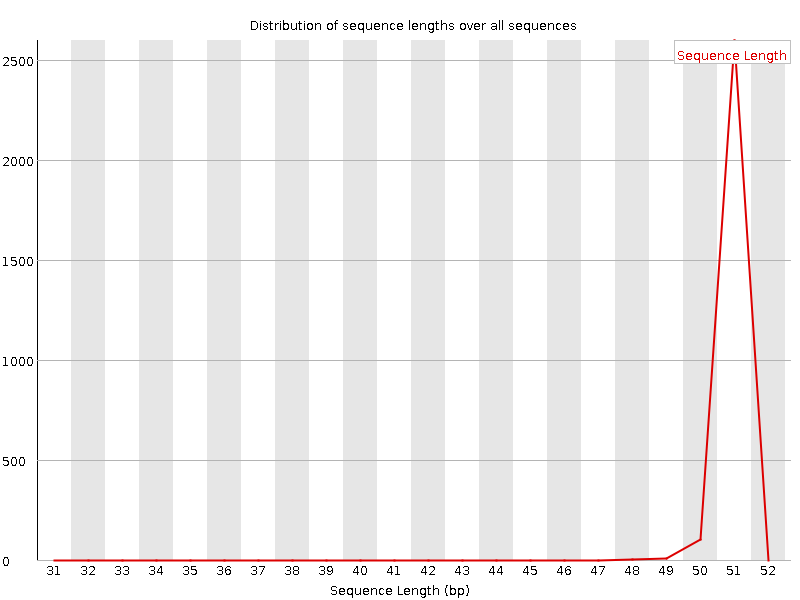
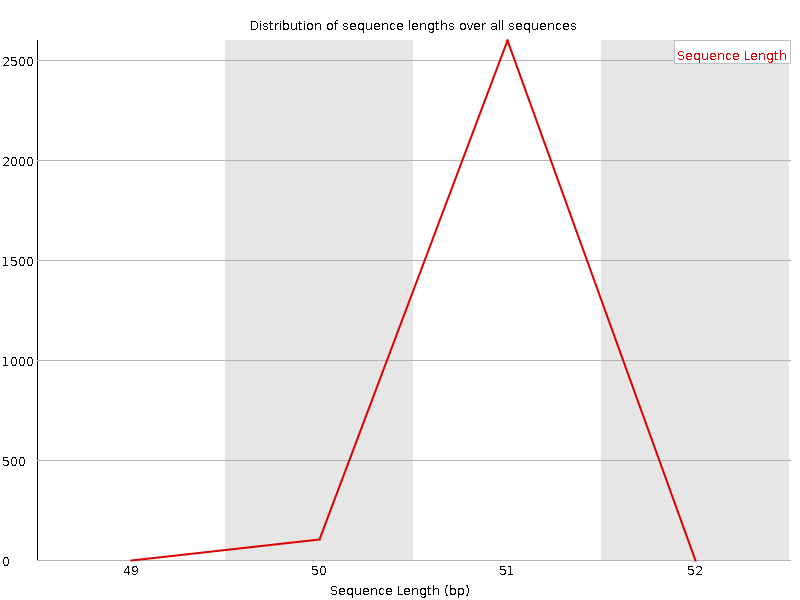
В рамках данного задания я также рассматривала 4 хромосому. Исходное число чтений составляло 2735, после чистки их осталось 2705. Судя по распределениям длины последовательностей, такое различие связано именно с отсеиванием слишком коротких чтений (рис. 1, 2). Также стоит заметить, что в данном материале повышенное содержание GC-пар - 52% как в исходном, так и очищенном варианте.
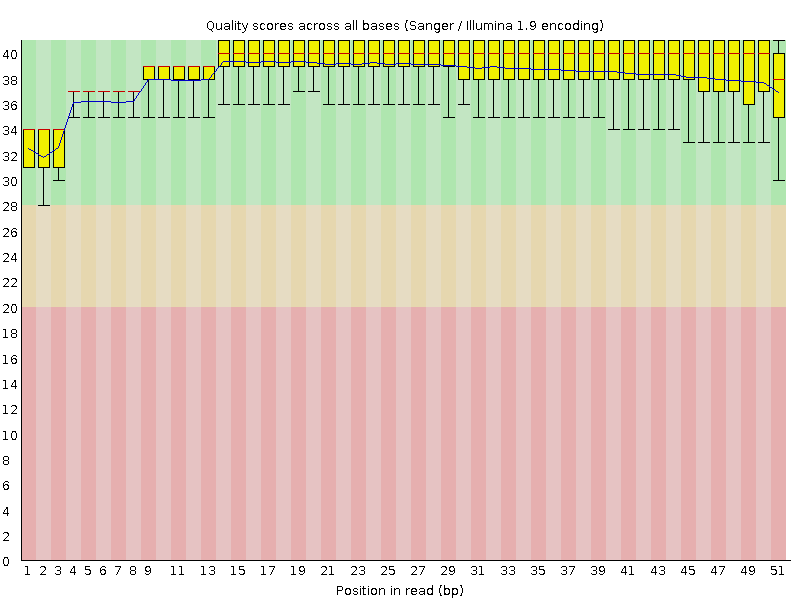
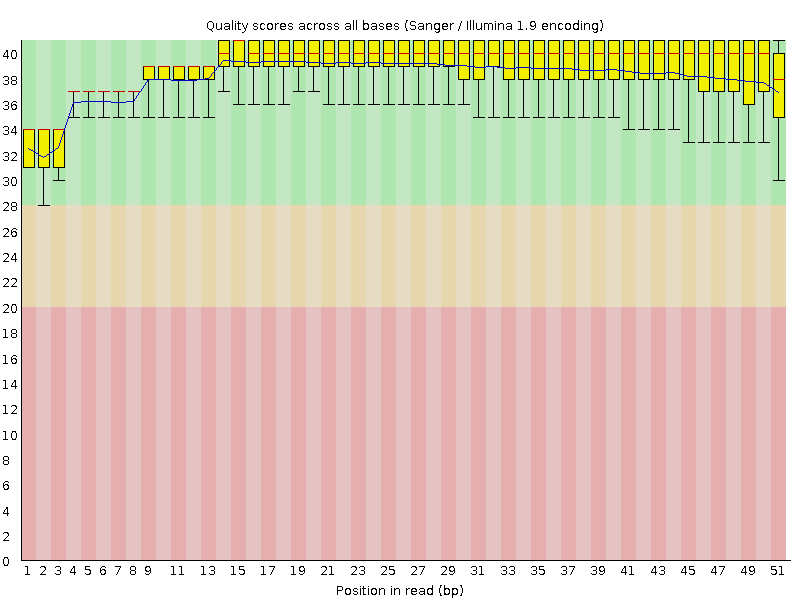
Качество самих ридов можно назвать хорошим, что иллюстрируют графики распределения качества до и после триммирования (рис. 3, 4).
| Команда | Назначение |
fastqc chr4_1.fastq | Анализ качества последовательности |
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr4_1.fastq chr4_1_trim.fastq TRAILING:20 MINLEN:50 | Очистка: обрезание ридов с качеством ниже 20 и длиной ниже 50 |
fastqc chr4_1_trim.fastq | Анализ качества после очистки |
hisat2 -x indexed -U chr4_1_trim.fastq -S chr4_1_align.sam --no-softclip | Картирование ридов. Был удален параметр --no-spliced-alignment, поскольку, используя его, мы скорее всего потеряли бы часть данных. В эукариотическом геноме огромную часть генома занимают интроны, которые удаляются из транскриптомных последовательностей путем сплайсинга. Как следствие, программе нельзя запрещать индели. Также было запрещено "подрезание" чтений. |
samtools view -b chr4_1_align.sam -o chr4_1_align.bam | Трансформация в бинарный формат |
samtools sort chr4_1_align.bam chr4_1_align_sorted | Сортировка выравниваний по координате RS |
samtools index chr4_1_align_sorted.bam | Индексирование выравнивания |
samtools flagstat chr4_1_align_sorted.bam > align_an.txt | Дополнительная информация о выравнивании |
htseq-count -s no -f bam -i gene_id chr4_1_align_sorted.bam index.gtf -o count_chr4_1.sam | Подсчет чтений; -s no- рассмотрение двух направлений цепи; -f bam- формат выходного файла; -i gene_id- задание индекса; -m(не указан - по умолчанию union) - "режим обработки чтений" - какое положение рида считать попаданием в ген. (другие флаги: intersection-strict и intersection-nonempty) |
Касаемо самого выравнивания - качество хорошее, о чем говорит выдача hisat2:
2705 reads; of these:
2705 (100.00%) were unpaired; of these:
72 (2.66%) aligned 0 times
2633 (97.34%) aligned exactly 1 time
0 (0.00%) aligned >1 times
97.34% overall alignment rate
С помощью фильтра и COUNTIF Excel была получена информация о попадании ридов в гены (таблица 2).
| Попадание | Число |
| ENSG00000261490.1 | 2 |
| ENSG00000071127.12 | 1861 |
| ENSG00000223086.1 | 1 |
| No feature | 769 |
| Not aligned | 72 |
Судя по всему, 769 последовательностей не попало непосредственно в ген. Это могло произойти из-за посттранскрипционных изменений, содержания промоторных областей, возможно даже ошибок полимераз. Первый ген в ensembl имеет пометку "sense overlapping". Пройдя по цепочке ссылок мы увидим, что он кодирует белок WDR1, по описанию сильно схожий со описанным ниже. Второй ген кодирует некий WD repeat-containing protein 1, который (в комплексе с другими белками) ответственен за разборку актиновых филаментов; участвует в цитокинезе и хемотаксической миграции клеток. Третий ген обозначен как "RNA, 5S ribosomal pseudogene 155", то есть, является псевдогеном.