Для выполнения задания получил файл chipseq_chunk46.fastq, содержащий риды Illumina одного из участков хромосомы человека. К сожалению, риды в нем были очень плохого качества, поэтому для работы я
взял резервный файл chipseq_chunk7.fastq. Сначала провёл
контроль качества ридов с помощью команды fastqc chipseq_chunk7.fastq. Результат выполнения команды - zip-архив - содержал файл fastqc_report.html,
в котором содержался отчёт о проделанной программой работе. Всего ридов 6543. Kачество чтения очень высокое на всём протяжении последовательности (видно на графике Per base sequence quality), поэтому
программа Trimmomatic для очистки чтений от участков плохого качества не применялась.
Риды были картированы на проиндексированный геном человека:
bwa mem /srv/databases/ngs/hg19/GRCh37.p13.genome.fa chipseq_chunk7.fastq > chipseq_chunk7.sam
Полученное выравнивание было проанализировано:
- Выравнивание переведено в бинарный формат:
samtools view -b chipseq_chunk7.sam -o aln.bam
- Отсортировано по координате в референсе начала чтения:
samtools sort aln.bam out.predix.bam
- Полученный файл проиндексирован:
samtools index out.predix.bam
- Получены данные о количестве откартированных ридов (см. рис. 2):
samtools idxstats out.predix.bam
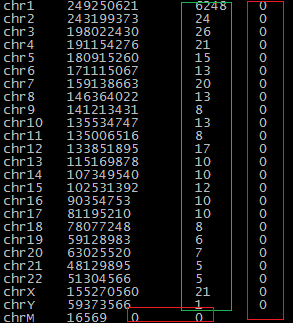 Рис.2. Число картированных ридов выделено ярко-зеленым, некартированных - красным.
Рис.2. Число картированных ридов выделено ярко-зеленым, некартированных - красным.
На хромосому картировались все риды без исключения, при этом на митохондриальную хромосому не картировалось ни одного рида; большинство ридов было картировано на первую хромосому. Следовательно, именно с
неё и были почитаны риды. Программа MACS была запущена со следующими параметрами:
macs2 callpeak -n chunk7 -t out.predix.bam --nomodel
На выход программа дала три файла:
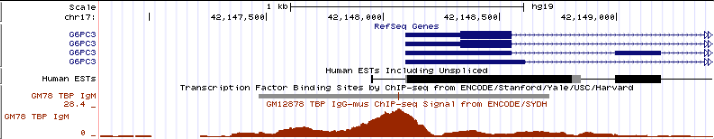
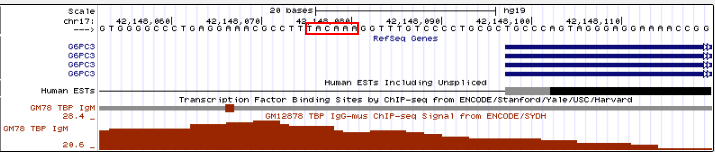
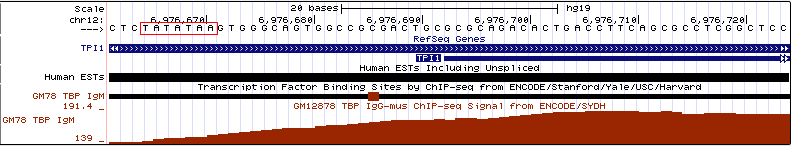
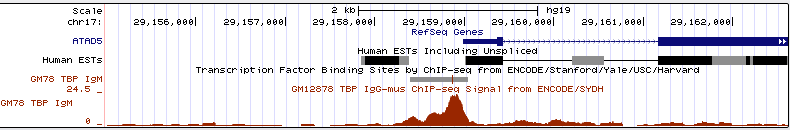
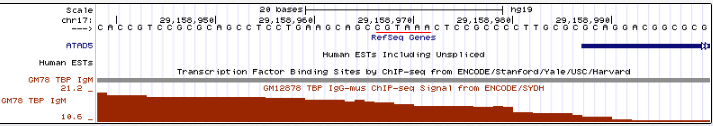
Всего программа нашла 9 пиков. Они были визуализированы в UCSC Genome Browser при помощи файла chunk7_peaks.narrowPeak. Для этого в него были добавлены следующие
строки:
track type=narrowPeak visibility=3 db=hg19 name="my_peaks" description="Peaks from chunk 7"
browser position chr1:59248597-59960068
Файл был подан на вход форме custom tracks в UCSC Genome Browser. Скриншот - на рис.3.:
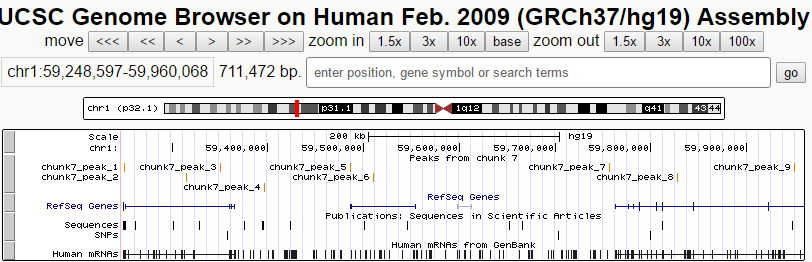 Рис.3. 10 пиков, открытые в Genome Browser.
Рис.3. 10 пиков, открытые в Genome Browser.
Рассмотрим подробнее пики 1 и 2:
| Пик | 1 | 2 |
| Длина пика | 478 | 200 |
| -LOG10(pvalue) | 53.41487 | 12.63990 |
| Вершина пика относительно начала | 168 от начала пика | 120 от начала пика |
| Положение в геноме | Пересекают экзон гена LINC01135, принадлежащий длинной некодирующей РНК 1135. |
Чем больше величина -LOG10(pvalue), тем меньше числовое значение pvalue и, следовательно, тем достовернее пик. Cледовательно, первый пик является самым достоверным из всех.