Общее описание системы
Силовое поле используемое при построении топологии системы - amber99sb
Заряд системы -10. ДНК отрицательно заряжена все-таки. Компенсируется 10 протонами.
Размер и форма ячейки: 5.01400nm *5.00700nm *5.26800nm - почти куб.
integrator = l-bfgs - алгоритм минимизации энергии.
coulombtype = Cut-off
vdwtype = Cut-off
Модель, которой описывался растворитель: fam.itp
Для биополимеров, укажите параметр который обуславливает неподвижность биополимера - define = -DPOSRES
Число шагов - nsteps = 10000
Длина шага - dt = 0.001 ps
Алгоритм расчёта электростатики - coulombtype = pme
и Ван-дер-Ваальсовых взаимодействий - vdwtype - cut-off
Алгоритмы термостата и баростата - Berendsen.
Длина траектории - 1 ps
Число шагов - 1000000
Длина шага. - 0.002 ps
Примечательно, что в файле написано total = 1.0 ps, хотя на самом деле итоговое время составляет 2.0 ps.
Алгоритм интегратора - md
Алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий - pme и Cut-off соотвественно
Алгоритмы термостата и баростата - v-rescale и Berendsen
1.pdb. Поскольку днк стремительными скачками перемещается по всему экрану, полученный результат был преобразован в fit.pdb. В общем-то результат схож - уже на 3-ем этапе (t=400) исчезает водородная связь между первой нуклеотидной парой, на 84 (t=16600) - у второй. После преобразования возник весьма забавный баг на 55 модели (t=10800) полного рассоединения до 68 модели за исключением 56, 60 и 63. Изначально я счел, что данный момент - ошибка алгоритма преобразования, так как в оригинальном pdb структура едина, но последующие исследование заставляют усомнится в том, что это просто ошибка.
Средне-квадратичное отклонение
Отклонение относительно стартовой структуры
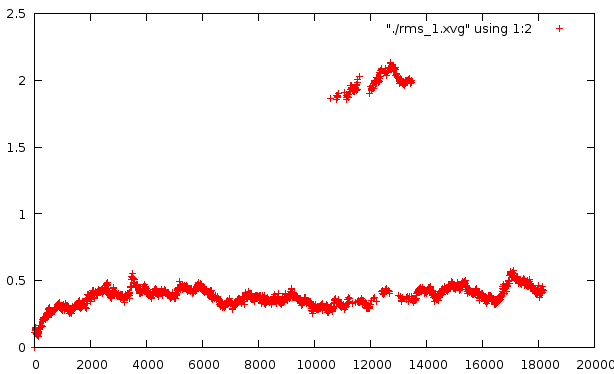
Отклонение на расстоянии 400 кадров
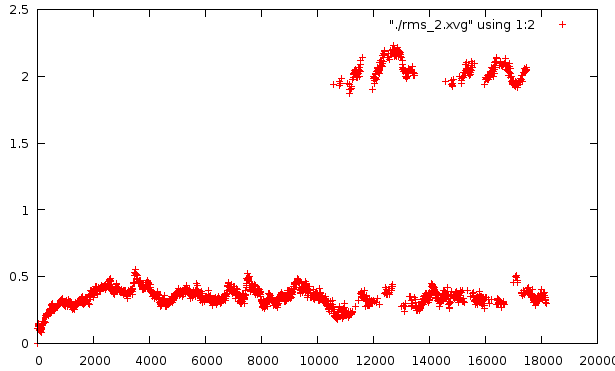
Отклонение на расстоянии 800 кадров
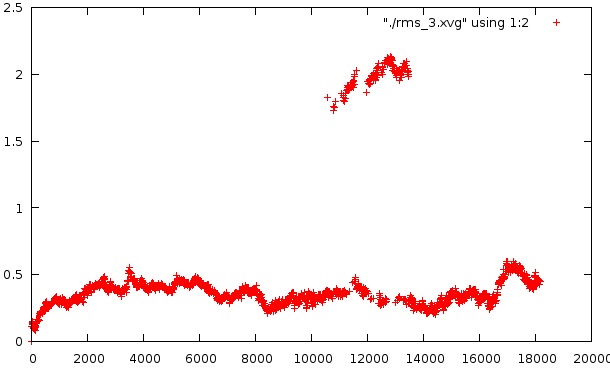
Отклонение на расстоянии 1200 кадров
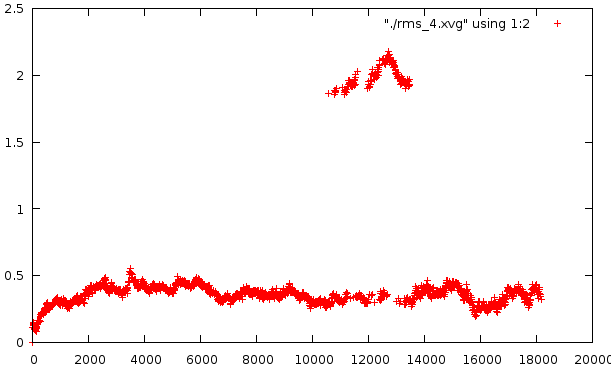
Отклонение на расстоянии 1600 кадров
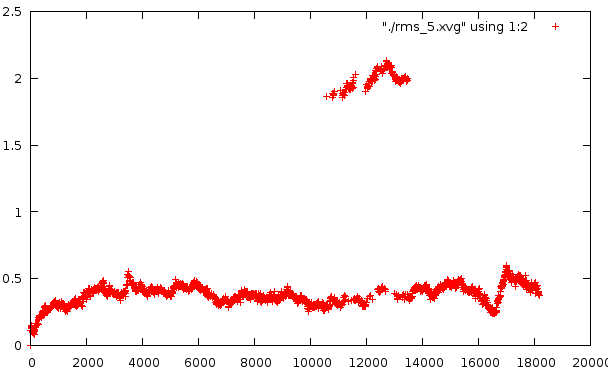
Любопытно само внезапное искажение в районе 10500-14000 - оно по временному отрезку сходится с тем. Второй график несет вторичное искажение, которое не отражено в структуре pdb. Наиболее низкое отклонение от базовой структуры наблюдается на графике №4, на графике №5 - самое мягкое искажение
О гидрофильности и гидрофобности

Колебания в изменении гидрофобной поверхности (красные кресты) незначительные. Однако гидрофильная поверхность изменяется, увеличиваясь в своих размерах - ведь молекула плавится, открывая все больше и больше гидрофильных зон. Резкий скачек идет на этап раскрытия первой водородной связи. Наблюдается странная неровность на графике на момент тех разрывов, и, наконец, бурный рост в конце на разрыве второй водородной связи
О водородных связях
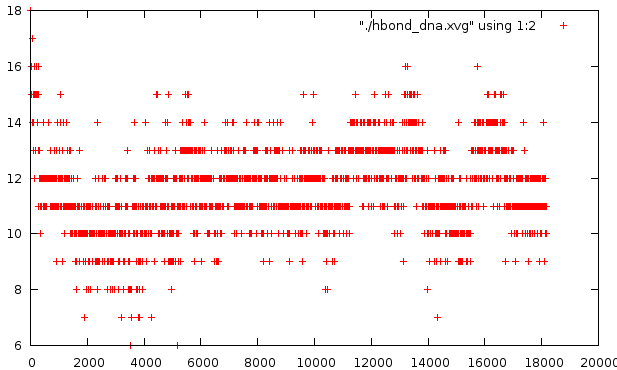
Изначально вдородных связей 14. Вообще плавление проходило довольно стабильно - наибольшее число связей за все время плавления было 10-13 - редко выпадало ниже, еще реже выходило вверх за этот предел.Этот график не подтверждает того искажения, что есть на втором варианте pdb - количество водородных связей не опускается ниже нуля
В итоге, можно сказать, что моделирование плавления ДНК не было завершено - внутренняя связь оказалась сильнее. Едва расщепившаяся связь в конце тому подтверждение. Очень спорен момент с полным расщеплением, что демонстрируется во втором pdb


Изменения модели с шагом в 0.02 пикосекунды - 1,06-1,12. Выглядит ОЧЕНЬ странно, ОЧЕНЬ неправдободобно. Подтверждается графиками о средне-квадратичном отклонении, опровергается графиком о гидрофильной поверхности (должен быть очень высокий пик, которого нет) и графиком о водородных связях (минимум водородных связей находится вообще не в этом временном отрезке). Резкое исчезновение и возобновление водородной связи (54-55, 56-57; 55-56 соответственно) действительно выглядит необычно.
Поскольку графики говорят об обратном, а то, что видят глаза (сравнивая 2 разных pdb) - неоднозначно, то в итоге все-таки я сделал вывод, что сиё полное расщепление - всего лишь ошибка обработки строчкой
trjconv -f dna_md.xtc -s dna_md.tpr -o dna_fit_1.pdb -skip 20 -fit rot+trans - ведь в оригинальном pdb (до обработки, чтобы не скакало) этого нет.