Изначально я выбрала домашнюю лошадь (Equus caballus), но из-за объёма её генома в итоге поменяла организм на Arabidopsis thaliana.
Сборка RefSeq: GCF_000001735.4
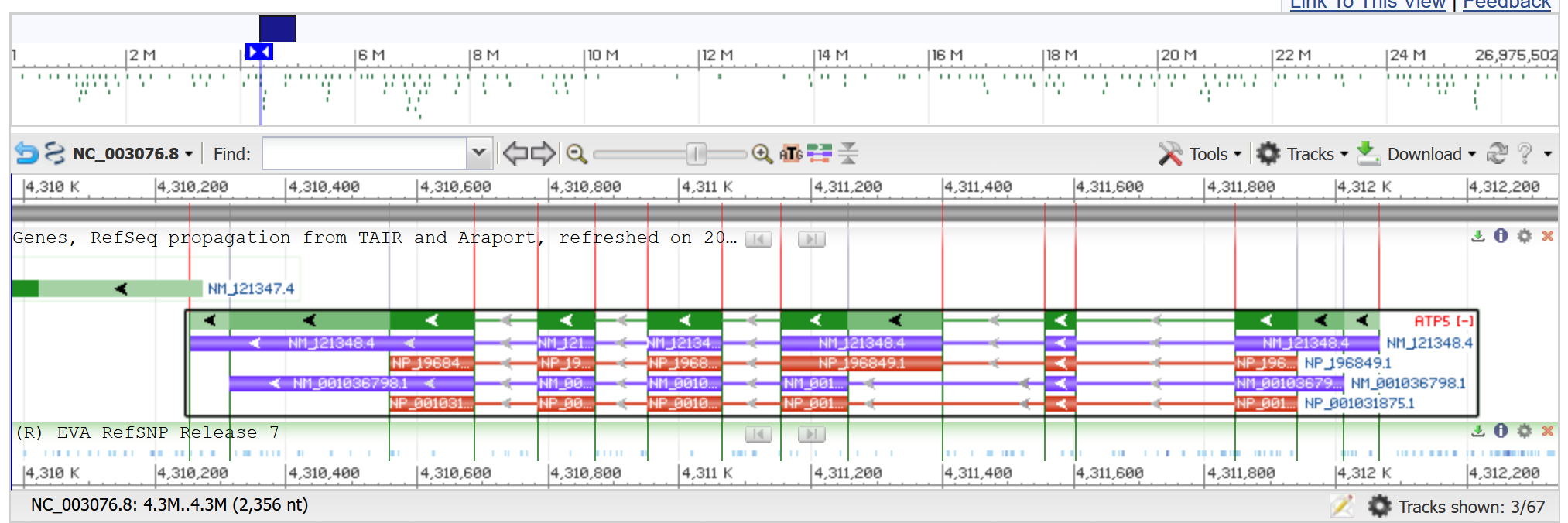
В задании требуется найти в геноме выбранного эукариота ген, кодирующий δ-субъединицу АТФ-синтазы
Последовательность белка, аннотированная как нужный ген была найдена с помощью текстового поиска. Её идентификатор: NP_196849.1
Идентификатор нуклеотидной записи, к которой относится ген: NC_003076.8
FASTA-файл нуклеотидной последовательностью
В задании требовалось выбрать для своего организма далёкое семейство из четырёх представленных. Так как мой организм является растением, можно было выбрать любой таксон. Выбранный таксон: Собачьи (Canidae)
Использованная база данных: RefSeq Genomes
Число сборок, входящих в таксон: 8
Длина слова: 5
Остальные параметры по умолчанию
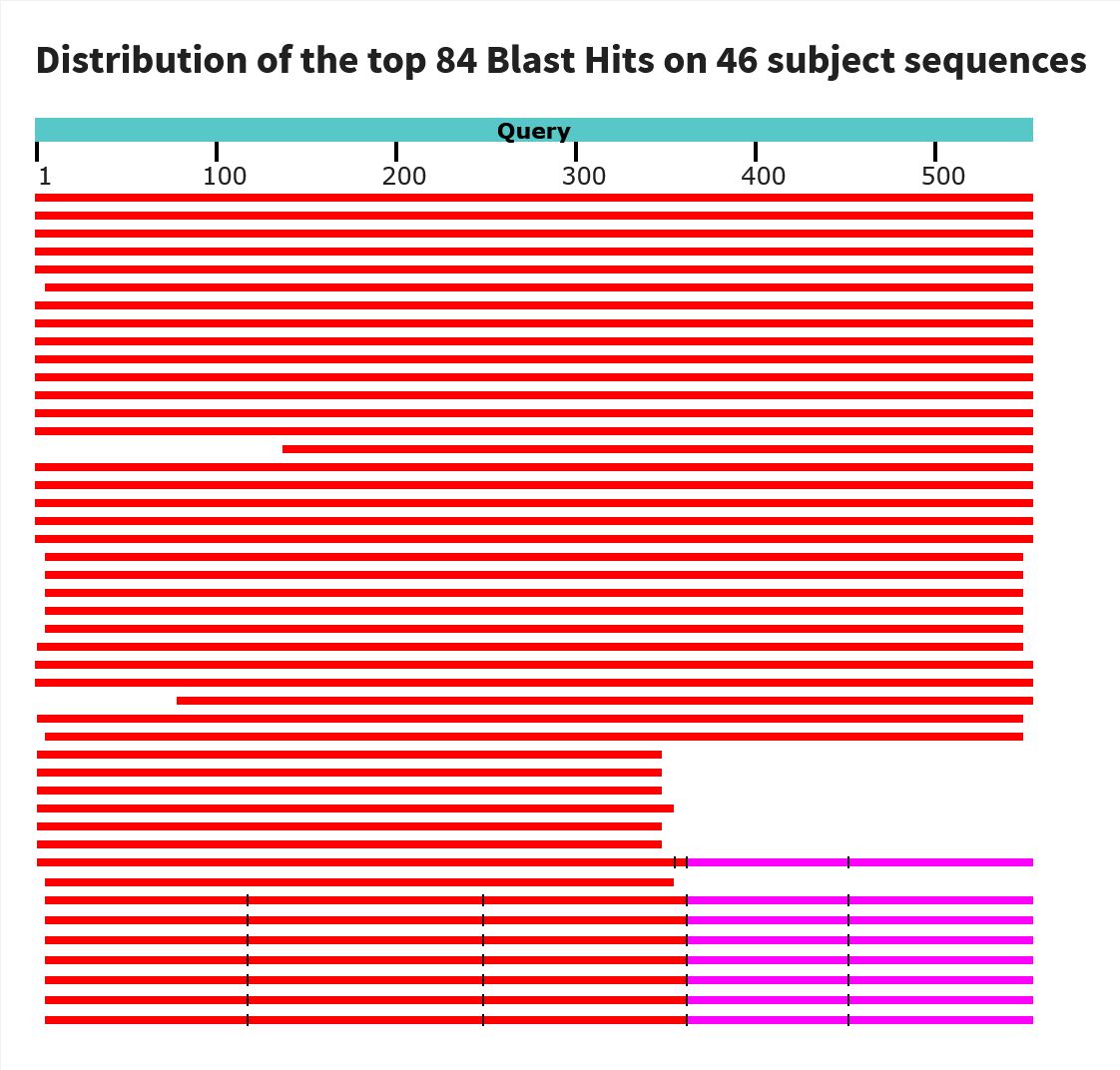
Однако, если проводить поиск по blastn, то blast не выдаёт результаты. Это показывает, что tblastn больше подходит для поиска гомологичных белков у отдалённых организмов. Чтобы решить проблему, я выбрала самую хорошую по E-value, покрытию и Per.Identity находку и использовала её для поиска в семействе Canidae с помощью алгоритма blastn
NC_135621.1 (28986859-28986308) - находка, по которой проводился поиск.
На локальном компьютере с помощью BLAST+ была проиндексирована последовательность Arabidopsis thaliana для последующей работы локального BLAST. Используемая команда:
makeblastdb -in GCF_000001735.4_TAIR10.1_genomic.fna -dbtype nucl -out anya.fna
-dbtype - указываем тип последовательности
После этого были скачаны последовательности рРНК (16S и 23S) Escherichia coli и проведен локальный поиск BLAST по полученной базе данных по методу blastn. Я выбрала этот метод, так как он хорошо подходит для поиска далёких последовательностей.
Команда для выполнения (16S): blastn -task blastn -query ./16s.fa -db ./anya.fna -outfmt 7 -evalue 0.005 -out 16sblast_anya
Алгоритм выдал 12 хитов, всего в шести последовательностях.
Команда для выполнения (23S): blastn -task blastn -query ./23s.fa -db ./anya.fna -outfmt 7 -evalue 0.005 -out 23sblast_anya
Алгоритм выдал 25 хитов, всего в пяти последовательностях
В выдаче видно, что первые находки для каждой из рРНК имеют очень хороший e-value.
Описание выдачи для 23S рРНК
| Accession сборки в Nucleotide | Описание AC сборки | Описание результата |
| NC_000932.1 | хлоропластный геном | две полных последовательности 23S рРНК |
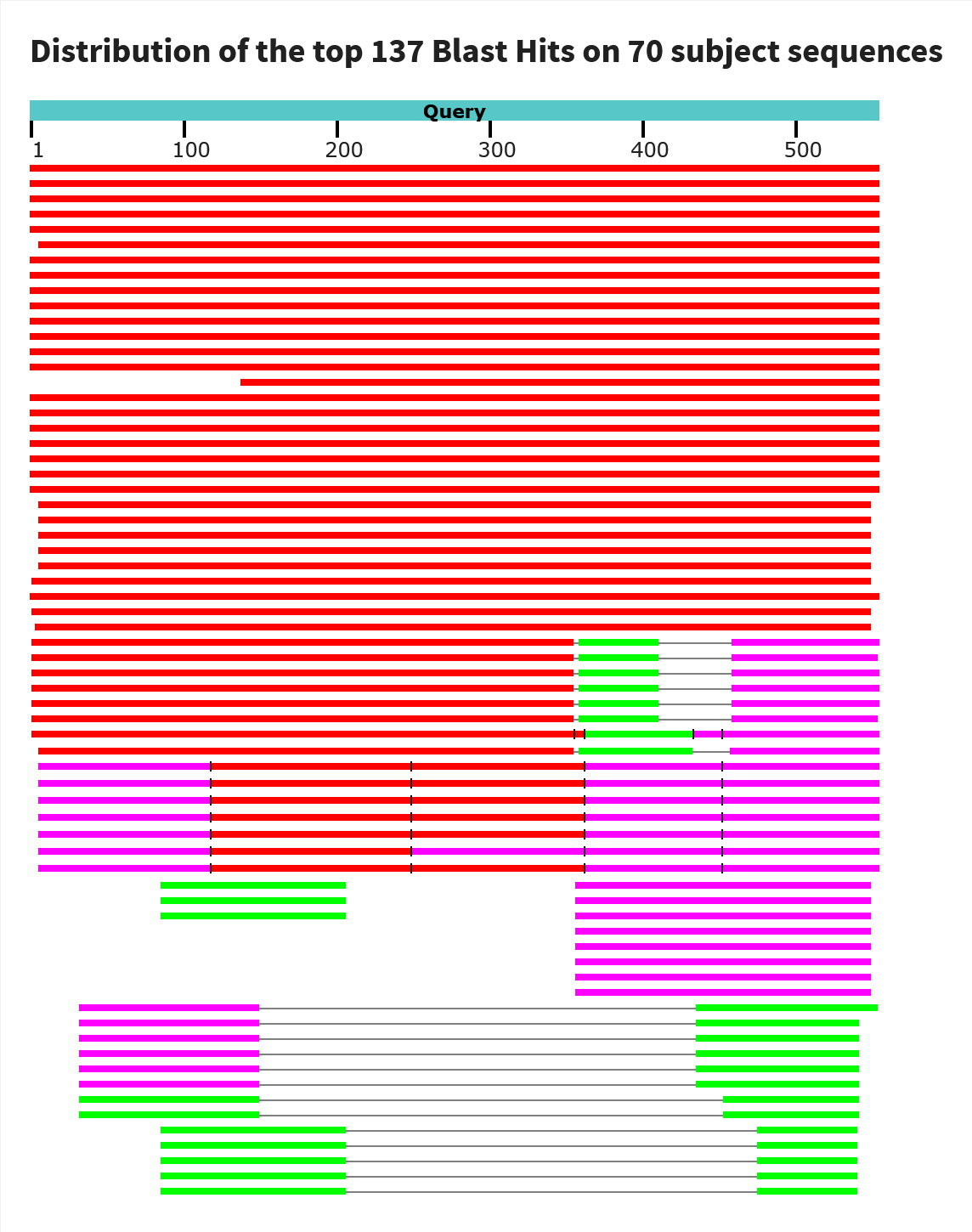
| NC_037304.1 | митохондриальный геном | гомологичные участки длиной 1093 и 978 на обратной цепи, полагаю, являются фрагментами одной копии, т.к. расстояние между ними всего ~400 нуклеотидов. Также на обратной цепи есть ещё один маленький участок длиной 67нт, но имеющий 83.582% идентичности. Думаю, это тоже может быть кусочком 23S рРНК. И есть ещё два участка гомологии на прямой цепи, тоже с высоким процентом идентичности. Итого: четыре копии 23S рРНК. |
| NC_003071.7 | 2 хромосома | на второй хромосоме есть две вставки 23S рРНК, одна из которых находится на обратной цепи и имеет длину около 150 нт, а другая, на прямой, разбита на четыре непересекающихся участка |
| NC_003070.9 | 1 хромосома | на прямой цепи есть шесть фрагментов одного и того же участка (2561-2621). Они разбиты на две группы по три, в одной группе участки располагаются относительно недалеко друг от друга. Видимо, произошла дупликация одного из кластеров. Также, копия практически такого же участка есть и на обратной цепи, но её e-value ниже, поэтому можно сказать, что она более древняя. Последний участок из тех восьми, что выдал бласт для этой хромосомы имеет самый низкий e-value и bit score, находится на прямой цепи |
| NC_003074.8 | 3 хромосома | все находки находятся на одной цепи, не пересекаются, общая длина чуть меньше 3000 нт, так что это ещё одна копия 23S рРНК |
Описание выдачи для 16S рРНК
| Accession сборки в Nucleotide | Описание AC сборки | Описание результата |
| NC_000932.1 | хлоропластный геном | две полных последовательности 23S рРНК на разных цепях |
| NC_037304.1 | митохондриальный геном | все пять находок на обратной цепи. При этом, копия 16S рРНК разбита на четыре части, а находка на координатах 269839-269789 является дупликацией одной из них (т.к. одинаковая длина гомологичного участка и bit score) |
| NC_003071.7 | 2 хромосома | всего две одинаковые находки, которые располагаются на разных цепях, но примерно в одном месте на хромосоме в целом. То есть, их координаты: 3250734-3250684 и 5464-5510. Думаю, это комплементарные последовательности, а копия одна |
| NC_003076.8 | 5 хромосома | маленькая находка на прямой цепи |
| NC_003074.8 | 3 хромосома | маленькая находка на прямой цепи |
| NC_003070.9 | 1 хромосома | маленькая находка на прямой цепи |
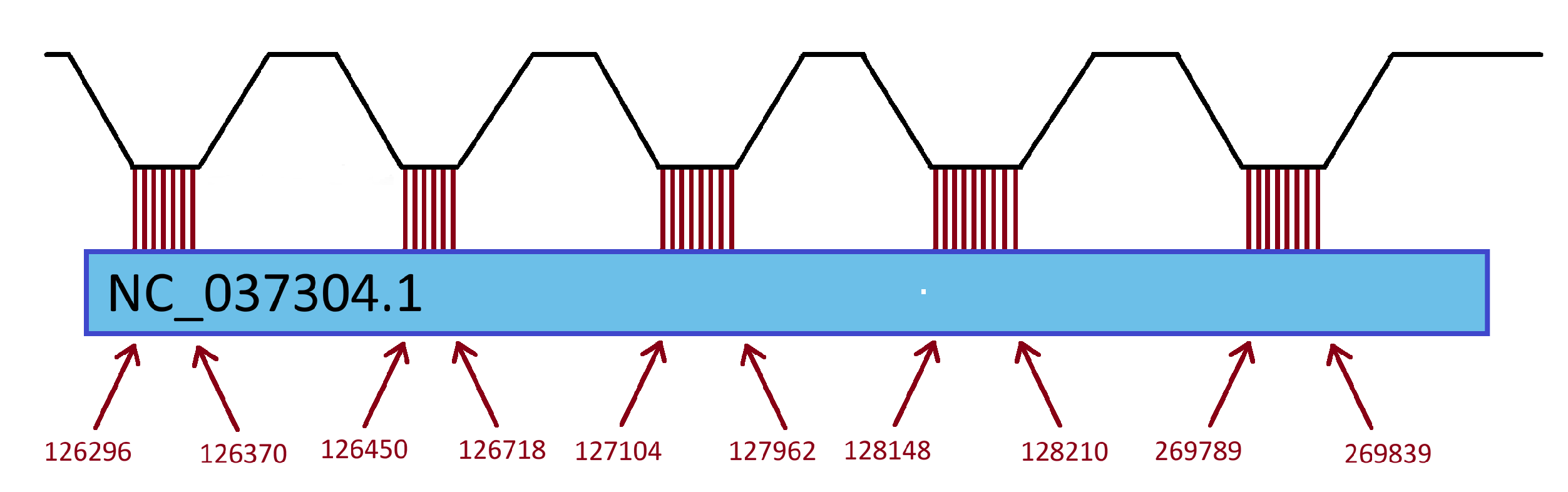
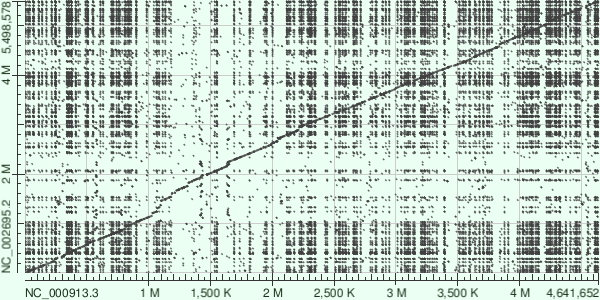
Для выполнения задания были выбраны два разных штамма кишечной палочки: Escherichia coli K-12 MG1655, Escherichia coli O157:H7 Sakai.
Диагональ посередине это большие консервативные блоки, сохранившиеся между штаммами. Много мелких делеций. Возможно есть транслокация в начале генома
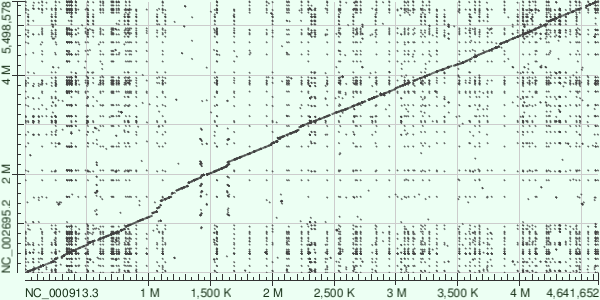
На карте BLASTN по всей площади графика есть большое количество мелких локальных совпадений. Это ожидаемо, так как blastn находит короткие участки гомологии. На главной диагонали видна линия сходства между двумя штаммами