Для реконструкции филогенетических деревьев использовалось "эталонное" выравнивание доменов семейства Asparaginase (точнее, его участок - см. benchmark.msf - в задании разрешается), полученное из базы данных Pfam (как это происходило - см. страничку"Сравнение фрагмента полного множественного выравнивания...". Вот:
| * | 2 | 0 | * | 4 | 0 | * | 6 | 0 | * | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| A | S | P | G | _ | M | Y | C | L | E | : | G | V | V | V | T | H | G | T | D | T | M | E | E | T | A | L | W | L | D | L | T | Y | A | G | - | N | V | P | V | V | L | T | G | A | M | R | S | A | D | A | P | N | A | D | G | P | T | N | L | R | E | A | L | A | V | A | A | S | P | A | A | R | G | V | G | V | L | V | S | F | : | 6 | 9 | ||||
| A | S | P | G | 2 | _ | Y | E | A | S | : | G | A | V | V | T | H | G | T | D | T | M | E | E | T | A | F | F | L | D | L | T | I | N | S | - | E | K | P | V | C | I | A | G | A | M | R | P | A | T | A | T | S | A | D | G | P | M | N | L | Y | Q | A | V | S | I | A | A | S | E | K | S | L | G | R | G | T | M | I | T | L | : | 6 | 9 | ||||
| A | S | P | G | 1 | _ | B | A | C | S | : | G | F | V | I | T | H | G | T | D | T | M | A | Y | T | S | A | A | L | S | Y | M | L | Q | H | A | K | K | P | I | V | I | T | G | S | Q | I | P | I | T | F | Q | K | T | D | A | K | K | N | I | T | D | A | I | R | F | A | C | E | G | - | - | - | V | G | G | V | Y | V | V | F | : | 6 | 7 | ||||
| A | S | P | G | 4 | _ | S | C | H | P | : | G | I | V | I | T | H | G | T | D | S | L | E | E | T | A | M | F | L | D | L | T | I | S | T | - | A | K | P | I | V | V | V | G | A | M | R | P | S | T | A | I | G | A | D | G | P | M | N | L | L | N | A | V | A | V | A | S | S | N | Q | S | M | G | R | G | T | L | V | L | L | : | 6 | 9 | ||||
| G | A | T | D | _ | M | E | T | K | A | : | G | V | V | I | G | H | G | T | D | T | M | A | F | T | A | A | A | L | S | F | V | I | E | G | L | N | G | P | V | V | L | V | G | A | Q | R | S | S | D | R | P | S | S | D | A | A | S | N | L | I | A | A | C | A | F | A | G | D | G | E | - | - | V | G | E | V | T | V | C | M | : | 6 | 8 | ||||
| G | V | H | G | T | D | T | L | P | G | D | N | A | A |
Для работы была создана Excel-книга UPGMA.xls.
Эволюционным расстоянием (D) считаем величину D = 100 – P, где P — процент идентичности. На основании матрицы попарных совпадений (см. "Практикум 10")
проводились расчеты; была создана матрица попарных эволюционных расстояний.
Результат - см. UPGMA.xls ; лист "Distances".
Результат - см. UPGMA.xls ; лист "UPGMA". Порядок действий - cогласно алгоритму UPGMA (см. "Подсказки..." - подробное описание):
Визуализация такая:
|
|
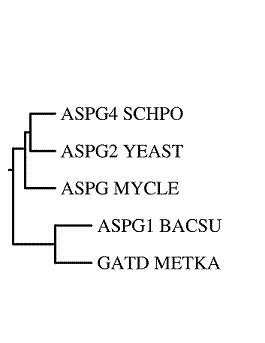
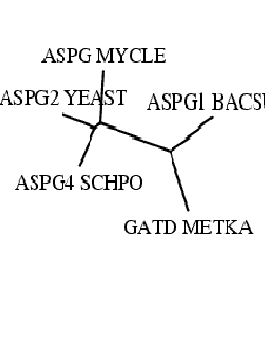
| Изображение, полученное drawgram. | Изображение, полученное drawtree. |
Программа drawgram построила укорененное дерево, а программа drawtree — неукорененное. Объективно, они дают представления о топологии, но не дают (связано это с работой алгоритма UPGMA (расстояния вычислялись как среднее арифметическое): "Алгоритм строит ультраметрическое дерево, а это означает, что скорость эволюции предполагается одинаковой для всех ветвей дерева. Использовать этот алгоритм имеет смысл только в случае ультраметрических данных (справедливости «молекулярных часов»)".) - о скоростях мутаций, "приведших" к формированию последовательностей. Да и насчет топологии - тоже следует уточнить:"помехой" может стать длина рассматриваемого участка выравнивания (малый объем выборки).
Лучше мое представление о данном дереве отражает вариант drawgram - укорененное дерево, т.к., строя правильную скобочную структуру в соответствии с алгоритмом UPGMA, мы "последовательно двигались" от листьев к корню (наличие корня и отражено в выдаче drawgram). Неукорененное дерево от drawtree в известной мере затрудняет кратко описать сценарий эволюции, т.к. по нему неясно, где может вообще находиться гипотетический общий предок.
Cценарий эволюции (по реконструкции drawgram): от гипотетической общей предковой последовательности домена (я считаю, что говорить при некоторых допущениях можно именно о последовательности домена. Обоснование - работа над заданием 12 практикума (выяснилось, что белки с доменом аспарагиназа - преимущественно однодоменные)) аспарагиназы произошли предковые последовательности двух типов. От одной из них поизошли последовательности домена аспарагиназы BACILLUS SUBTILIS (ASPG1_BACSU) и METHANOPYRUS KANDLERI (GATD_METKA) ; от другой - последовательность домена аспарагиназы MYCOBACTERIUM LEPRAE (ASPG_MYCLE) и предковая последовательность домена аспарагиназы SCHIZOSACCHAROMYCES POMBE(ASPG4_SCHPO) и SACCHAROMYCES CEREVISIAE (ASPG2_YEAST). В свою очередь, эта предковая последовательность - последняя из вышеназванных - дала начало соответствующим последовательностям SACCHAROMYCES CEREVISIAE и SCHIZOSACCHAROMYCES POMBE.
Примечания (информация по систематике; получена через поисковую систему SRS - http://srs.ebi.ac.uk/):
BACILLUS SUBTILIS (систематика: BACTERIA; FIRMICUTES; BACILLALES; BACILLACEAE; BACILLUS)
METHANOPYRUS KANDLERI (систематика: ARCHAEA; EURYARCHAEOTA; METHANOPYRI; METHANOPYRALES; METHANOPYRACEAE; METHANOPYRUS)
MYCOBACTERIUM LEPRAE (BACTERIA; ACTINOBACTERIA; ACTINOBACTERIDAE; ACTINOMYCETALES; CORYNEBACTERINEAE; MYCOBACTERIACEAE; MYCOBACTERIUM)
SCHIZOSACCHAROMYCES POMBE (EUKARYOTA; FUNGI; ASCOMYCOTA; SCHIZOSACCHAROMYCETES; SCHIZOSACCHAROMYCETALES; SCHIZOSACCHAROMYCETACEAE; SCHIZOSACCHAROMYCES) и SACCHAROMYCES CEREVISIAE (EUKARYOTA; FUNGI; ASCOMYCOTA; SACCHAROMYCOTINA; SACCHAROMYCETES; SACCHAROMYCETALES; SACCHAROMYCETACEAE; SACCHAROMYCES)
SACCHAROMYCES CEREVISIAE (EUKARYOTA; FUNGI; ASCOMYCOTA; SACCHAROMYCOTINA; SACCHAROMYCETES; SACCHAROMYCETALES; SACCHAROMYCETACEAE; SACCHAROMYCES)
Результат визуализации:
 |
 |
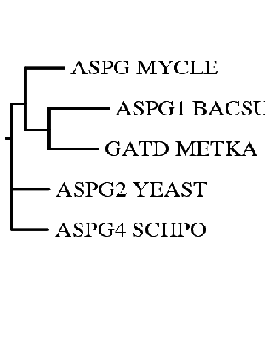
| Изображение, полученное drawgram (укорененное дерево... Филограмма). |
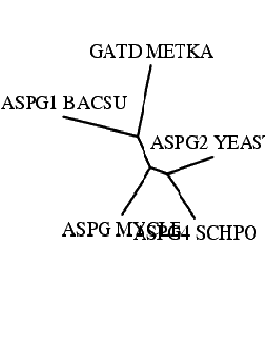
Изображение, полученное drawtree (неукорененное дерево). |
Итак, построение дерева методом ближайших соседей (neighbor-joining) произведено. Т.к. метод строит неукорененное дерево, то оптимальной
является визуализация drawtree (неукорененное дерево - его надо понимать как множество всех возможных укоренений).
Поэтому и стратегию, в данном случае, разумно приводить по неукорененному дереву. В свою очередь, неукорененные NJ-дерево и UPGMA-дерево
фактически оказываются одинаковыми: одинаково взаимное расположение ветвей; про длину ветвей речь не идет (алгоритмы ведь разные).
Что касается укорененных NJ-дерева и UPGMA-дерева, то здесь есть некоторое расхождение (а именно, более раннее "отщепление" последовательности ASPG_MYCLE).
Ответ на ГЛАВНЫЙ вопрос: при реконструкции деревьев разными методами в целом (речь идет о топологии - по "временным" показателям (скорость накопления мутаций в различных систематических группах) сравнивать некорректно /алгоритмы-то разные/) сценарий эволюции не изменился.